Clostridium botulinum: Difference between revisions
Jump to navigation
Jump to search
| Line 9: | Line 9: | ||
chromosome: 3,886,916 bp 28.24 GC% | chromosome: 3,886,916 bp 28.24 GC% | ||
plasmid: 16,344 bp 26.80 GC% | plasmid: 16,344 bp 26.80 GC% | ||
[https://wiki.umiacs.umd.edu/cbcb/images/3/31/CB-CB.png Mummerplot Complete Genome vs Complete Genome] | |||
* [ftp://ftp.sanger.ac.uk/pub/pathogens/cb/CB_shotgun.dbs Traces] | * [ftp://ftp.sanger.ac.uk/pub/pathogens/cb/CB_shotgun.dbs Traces] | ||
Revision as of 14:53, 9 August 2007
Data sources
Sanger:
Hall strain A (ATCC 3502) chromosome: 3,886,916 bp 28.24 GC% plasmid: 16,344 bp 26.80 GC%
Mummerplot Complete Genome vs Complete Genome
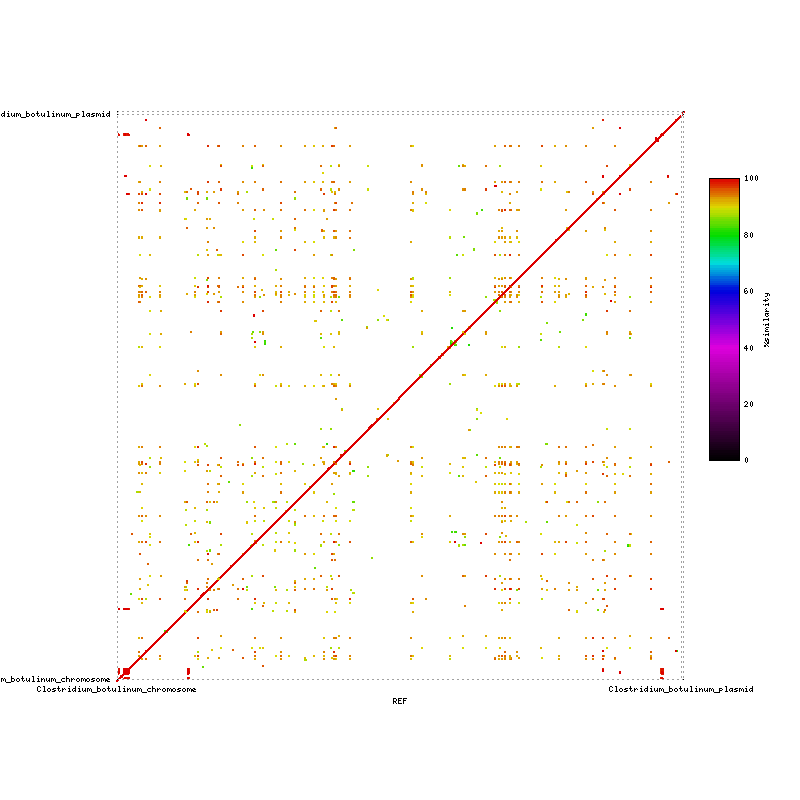{kind=link}
63,115 Sanger reads
Read problems:
no quality : default 20 assigned to all the bases
no mate pairing : can be inferred from names (.p1c, .q1c => 27,331 mates)
no library info : assumed there was only one library used
no trimming info : almost all reads have "CONTAINED" alignments to the reference
CLR=1,len(read)
there are 124 regions in the reference which are not covered by reads
NCBI :
Reads have not been submitted to TA
The initial genome assembly was obtained from 69,632 paired end sequences (giving 9.15-fold coverage) derived from four genomic shotgun libraries (all in pUC18 with insert sizes of 1.5–2.0 kb and 2.0–2.2 kb, 2.2–2.5 kb, and 2.5–4.0 kb) using dye terminator chemistry on ABI3700 automated sequencers; 1604 pairedend sequences from one pBACe3.6 library with insert sizes of 15–23 kb (a clone coverage of 3.9-fold) were used as a scaffold. A further 9343 directed sequencing reads were generated during finishing.
Assembly
Location:
/fs/szasmg/Bacteria/C_botulinum
- WGA
create a .frg file runCA-OBT.pl (default params) location: 2007_0725_WGA => 109 scaffolds, 243 contigs => library inser estimates mean=1840.917 stdev=866.039
- AMOScmp
MINCLUSTER=30 , MAXTRIM=50 location: 2007_0801_AMOScmp-relaxed => 2 scaffolds, 148 contigs CB.qc CB.chromo.png CB.plasmid.png CB-scaff.png
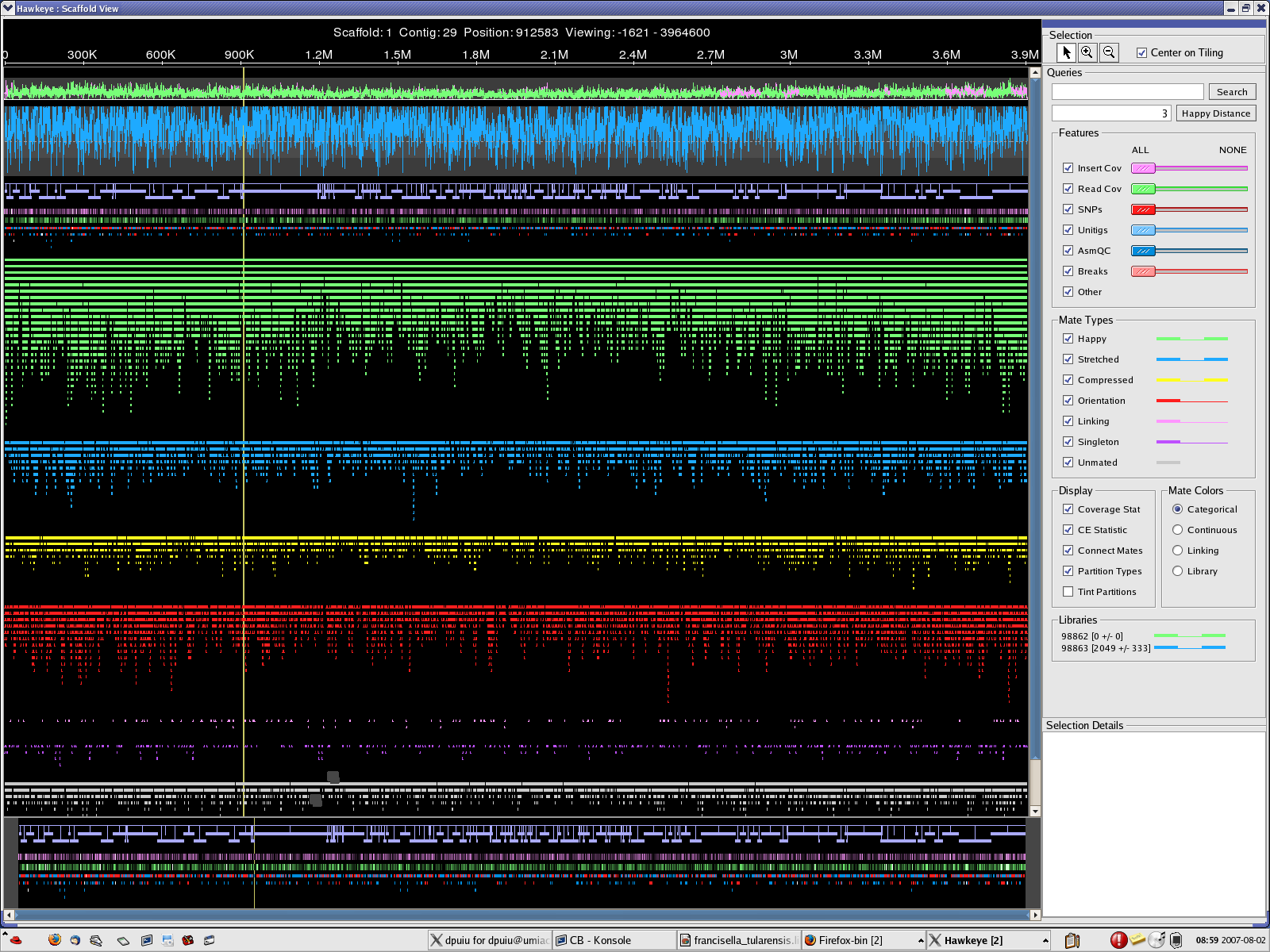{kind=link}
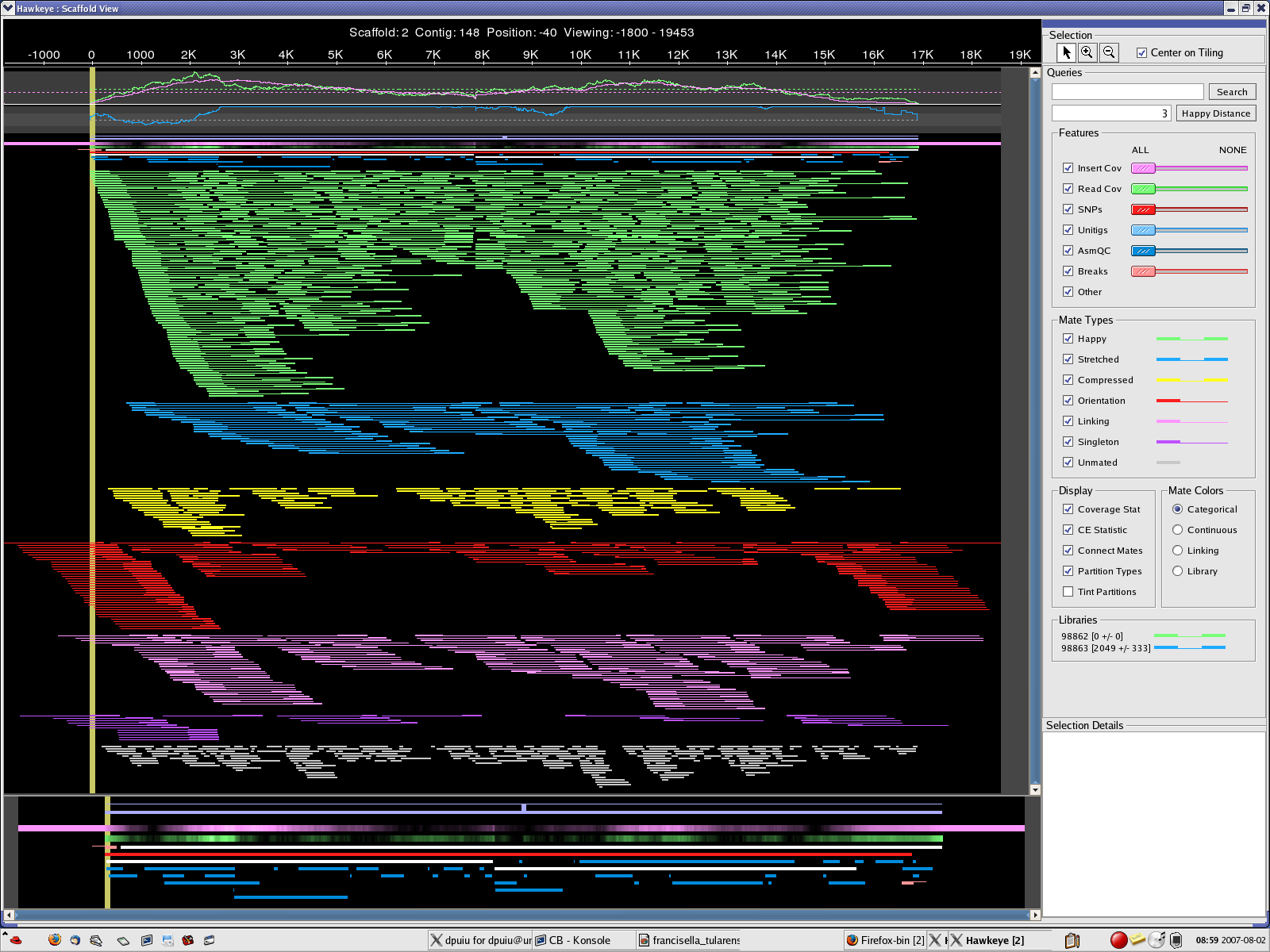{kind=link}
{kind=link}