Pseudodomonas syringae: Difference between revisions
Jump to navigation
Jump to search
No edit summary |
|||
| Line 62: | Line 62: | ||
== Assemblies == | == Assemblies == | ||
=== 454 + Solexa === | |||
/fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Solexa-454/2007_1009_AMOSCmp-relaxed | /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Solexa-454/2007_1009_AMOSCmp-relaxed | ||
142 contigs (37 negative gaps, 89 positive gaps) | 142 contigs (37 negative gaps, 89 positive gaps) | ||
| Line 80: | Line 76: | ||
'''454 single reads + 25 bp Solexa reads ''' => 293 contigs, 144 negative gaps, 131 positive gaps | '''454 single reads + 25 bp Solexa reads ''' => 293 contigs, 144 negative gaps, 131 positive gaps | ||
=== 454 + Solexa +454p === | |||
Only the 454 paired ends that contain 1 single complete adaptor sequence were used (allmost all) | Only the 454 paired ends that contain 1 single complete adaptor sequence were used (allmost all) | ||
/fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Solexa-454-454p/2007_1011_AMOSCmp-relaxed-filtered | /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Solexa-454-454p/2007_1011_AMOSCmp-relaxed-filtered | ||
149 contigs; very similar to the prev ome | 149 contigs; very similar to the prev ome | ||
3. | 3. === 454 + Solexa +454p === | ||
MAJORITY=50 | |||
/fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Solexa-454/2007_1015_AMOSCmp-relaxed-MAJORITY50 | /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Solexa-454/2007_1015_AMOSCmp-relaxed-MAJORITY50 | ||
131 contigs (18 negative gaps) | 131 contigs (18 negative gaps) | ||
| Line 130: | Line 126: | ||
ftp://ftp.cbcb.umd.edu/pub/data/dpuiu/Pseudomonas_syringae/Solexa-454 | ftp://ftp.cbcb.umd.edu/pub/data/dpuiu/Pseudomonas_syringae/Solexa-454 | ||
=== Sanger AMOScmp === | |||
'''Sanger reads''' | '''Sanger reads''' | ||
/fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1011_AMOSCmp-relaxed | /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1011_AMOSCmp-relaxed | ||
| Line 138: | Line 134: | ||
[[Media:Pseudodomonas_syringae.Sanger.AMOSCmp.chromosome_problem.png|Chromosome problem]] | [[Media:Pseudodomonas_syringae.Sanger.AMOSCmp.chromosome_problem.png|Chromosome problem]] | ||
=== Sanger Celera 3.11 === | |||
/fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1011_WGA | /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1011_WGA | ||
22 scaff, 46 contigs, 181 degens | 22 scaff, 46 contigs, 181 degens | ||
| Line 158: | Line 153: | ||
4898971 4955870 | 175592 118697 | 56900 56896 | 99.98 | 6397126 175592 | 0.89 32.40 | gi|28867243|ref|NC_004578.1| 7180000001443 | 4898971 4955870 | 175592 118697 | 56900 56896 | 99.98 | 6397126 175592 | 0.89 32.40 | gi|28867243|ref|NC_004578.1| 7180000001443 | ||
=== Sanger AMOScmp (Chromosome+3 plasmids ref) === | |||
Reference=complete genome(chromosome+3 plasmids) use "circular contig" in Celera 3.11 assembly | Reference=complete genome(chromosome+3 plasmids) use "circular contig" in Celera 3.11 assembly | ||
/fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1012_AMOSCmp-relaxed-3plasmids | /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1012_AMOSCmp-relaxed-3plasmids | ||
| Line 165: | Line 160: | ||
The missoriented read pile corresponding to the chromosome (4. AMOSCmp of Sanger reads) has dissapeared | The missoriented read pile corresponding to the chromosome (4. AMOSCmp of Sanger reads) has dissapeared | ||
AA ready for submission: /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1012_AMOSCmp-relaxed-3plasmids/AA/umd-20071030-141700.tar.gz | AA ready for submission: /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1012_AMOSCmp-relaxed-3plasmids/AA/umd-20071030-141700.tar.gz | ||
=== Solexa AMOScmp === | |||
1. align all reads (Solexa) to the reference using nucmer. I initially used minmatch=20, mincluster=20 (-c 20 -l 20) | |||
6340136 reads | |||
5641782 (88.98%) aligned by nucmer -c 20 -l 20 | |||
3453618 (54.47%) aligned by nucmer -c 32 -l 20 | |||
2707005 (42.69%) aligned by nucmer -c 32 -l 32 | |||
Revision as of 17:37, 20 February 2008
Pseudomonas syringae pv. tomato str. DC3000
Data
Originally sequenced and finished at TIGR: published Sept 2003
NCBI
AA: no assembly TA 80,959 reads Genome Project Taxonomy TaxId=223283
Chromosome + 2 plasmids:
Name Length %GC Info NC_004578.1 6,397,126 58.40 chromosome NC_004633.1 73,661 55.15 plasmid pDC3000A NC_004632.1 67,473 56.17 plasmid pDC3000B total 6,538,260
Little similarity between the chromosome and plasmids. The 2 plasmids share a significant amount of DNA; see /fs/szasmg2/Bacteria/Pseudomonas_syringae/Data/nucmer/NC_004633-NC_004632.png
UNC: Jeff Dangl
New sequence:
Read stats
Type File #reads min median max sum mean stdev n50 Solexa DC3000.reads.filtered.fasta 6,340,136 32 32 32 202884352 32 0 32 454p DC3000.format.454Reads.fna 123,992 38 86 329 15623908 126.01 58.89 142 454 DC3000.TCA.454reads.format.fna 77,466 35 244 371 18627363 240.46 26.85 245
* Solexa 3 lanes;
* 454 shotgun 1/4 Plate (250bp read);
* 454 paired ends 1/4 Plate :
* contain a 44 bp linker in the middle
* the linker sequence is: GTTGGAACCGAAAGGGTTTGAATTCAAACCCTTTCGGTTCCAAC
* there are some (not many) 454 paired end sequences that contain multiple instances of the linker (tandem): Example EUEIEUN01ANUGL_length=128_xy=0154_1891
Quality values are missing for all data sets!!!
I assigned default qual=3 to all the base (.frg & .afg files)
UNC sequence data: (not avail any more?)
http://biology622.dhcp.unc.edu/~labweb/DCData/
UNC (e-mail):
* Theoretical minimum number of contigs we can obtain is 268 (our reads fail to cover 269 nucleotides). * Our de novo assembly spans the genome in 853 contigs totaling 6,313,026 bp. * 98.7% of the genome is covered by a contig; * 84% of the genome is covered by contigs 10,000 bp or greater. * The average gap size between contigs is 98 bp; * average contig size 7401 bp. * The N50 = 37,444 bp. * Our largest BAMBUS "scaffold" is 2,565,761 bp
Files location:
/fs/szasmg2/Bacteria/Pseudodomonas_syringae/Data /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly
Assemblies
454 + Solexa
/fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Solexa-454/2007_1009_AMOSCmp-relaxed 142 contigs (37 negative gaps, 89 positive gaps) No read trimming was done. AMOScmp used the following parameters: nucmer -c 20 casm-layout -t 20 -o 5 "-t 20" allows for 20 bp long dirty sequence ends which seem to solve the "low quality" problem. => 22 large contigs 454 single reads + 30 bp Solexa reads => 167 contigs , 49 negative gaps, 100 positive gaps 454 single reads + 25 bp Solexa reads => 293 contigs, 144 negative gaps, 131 positive gaps
454 + Solexa +454p
Only the 454 paired ends that contain 1 single complete adaptor sequence were used (allmost all) /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Solexa-454-454p/2007_1011_AMOSCmp-relaxed-filtered 149 contigs; very similar to the prev ome
3. === 454 + Solexa +454p === MAJORITY=50
/fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Solexa-454/2007_1015_AMOSCmp-relaxed-MAJORITY50 131 contigs (18 negative gaps) No read trimming was done. AMOScmp used the following parameters: nucmer -c 20 casm-layout -t 20 -o 5 -m 50 No read trimming was done. "-t 20" allows for 20 bp long dirty sequence ends which seem to solve the "low quality" problem. "-m 20" merges some contigs together => 10 large contigs
contig# len gc% 4 2290968 59.00 7 1817904 58.18 3 1405326 58.08 5 648413 58.48 2 192413 57.86 6 87152 58.02 131 71251 56.47 1 32939 54.86 130 29120 59.36 9 20309 53.56 95 3589 59.46
Rerun Solexa32,Solexa30,Solexa25 with "nucmer -b 2 -g 5"
2007_1015_AMOSCmp-relaxed-Solexa32/
2007_1015_AMOSCmp-relaxed-Solexa30/
2007_1015_AMOSCmp-relaxed-Solexa25/
/fs/szasmg2/Bacteria/Pseudomonas_syringae/Assembly/Solexa-454/qc.combine.3
$ show-coords 1con-contigs.delta | grep gi | awk '{print $7}' | getSummary.pl # sum of ref alignments: 13608985
$ show-coords 1con-contigs.delta | grep gi | awk '{print $8}' | getSummary.pl # sum of qry alignments: 13747738
138,753 bp in duplications for Solexa32 ???
61,741 bp in duplications for Solexa30 ???
10,881 bp in duplications for Solexa25 ???
Copy of assembly files: /fs/ftp-cbcb/pub/data/dpuiu/Pseudomonas_syringae ftp://ftp.cbcb.umd.edu/pub/data/dpuiu/Pseudomonas_syringae/Solexa-454
Sanger AMOScmp
Sanger reads /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1011_AMOSCmp-relaxed Many miss-oriented mates in the 4.8M-5M region of the chromosome 22 contigs Chromosome Chromosome problem
{kind=link}
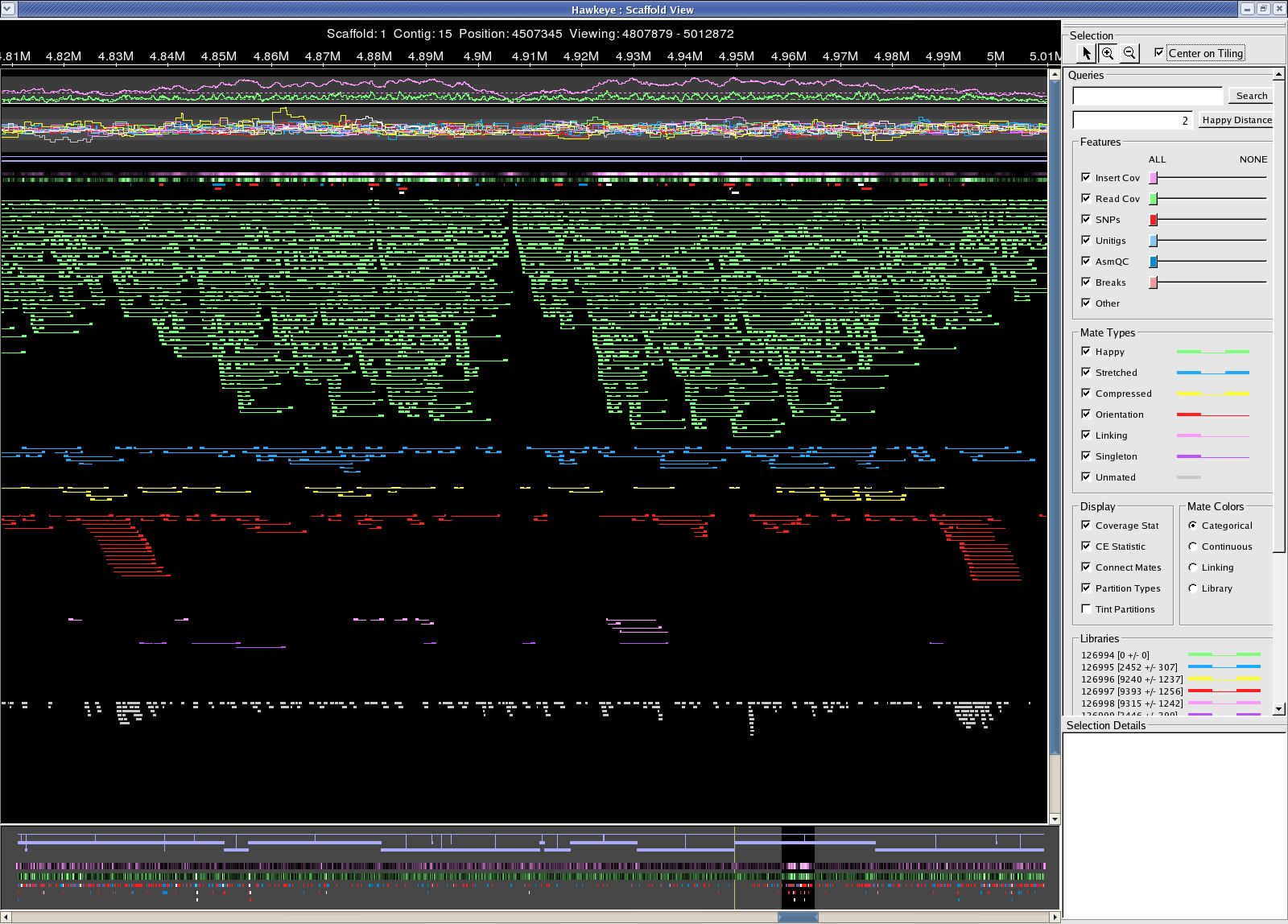{kind=link}
Sanger Celera 3.11
/fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1011_WGA 22 scaff, 46 contigs, 181 degens Scaffold 7180000001443 looks circular: possible 163,074 bp plasmid aligns to 4.8M-5M "problem" region in the chromosome 7180000001443.png
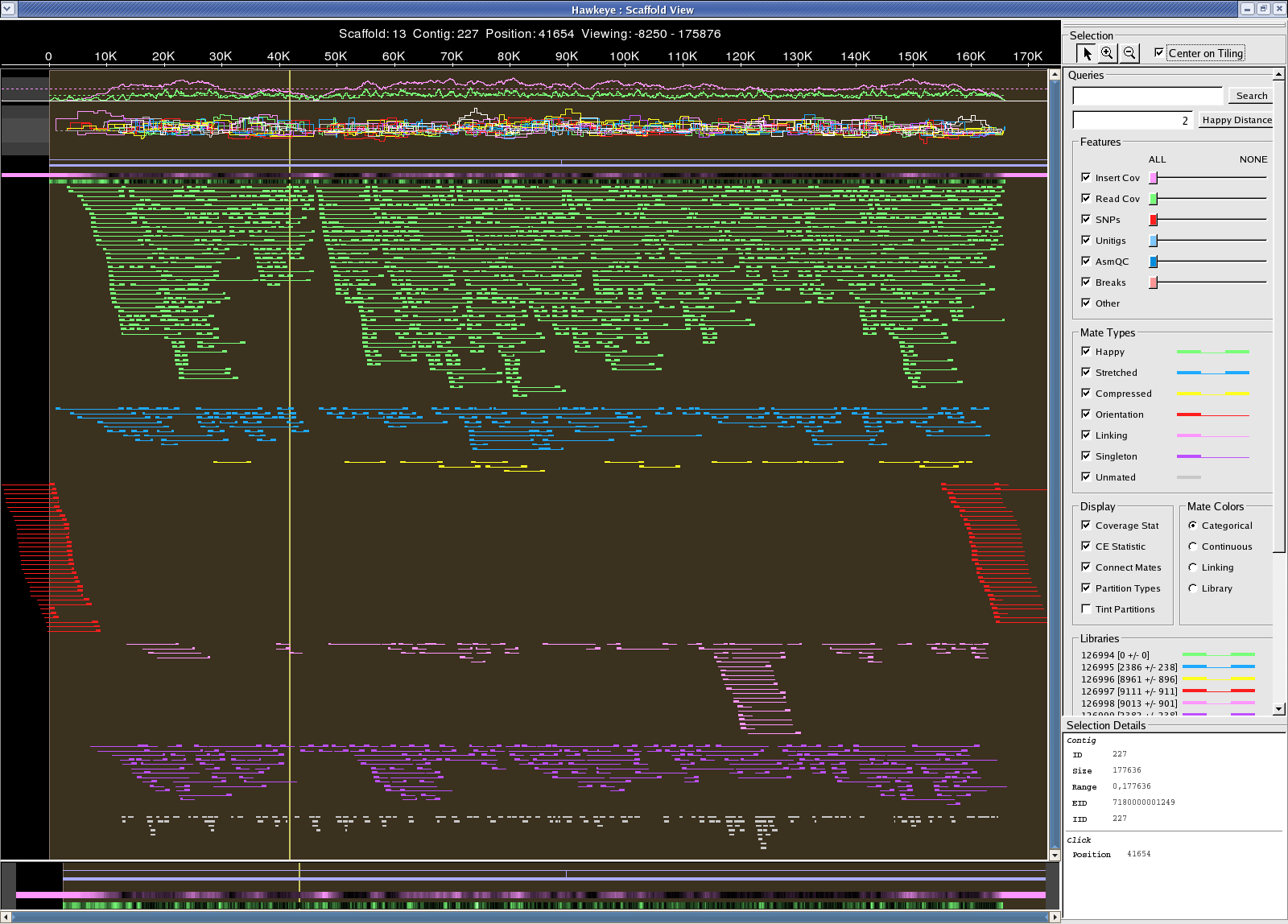{kind=link}
[S1] [E1] | [S2] [E2] | [LEN 1] [LEN 2] | [% IDY] | [LEN R] [LEN Q] | [COV R] [COV Q] | [TAGS]
===============================================================================================================================
1 175592 | 1 175592 | 175592 175592 | 100.00 | 175592 175592 | 100.00 100.00 | 7180000001443 7180000001443 [IDENTITY]
1 12519 | 163075 175592 | 12519 12518 | 99.98 | 175592 175592 | 7.13 7.13 | 7180000001443 7180000001443 [BEGIN]
163075 175592 | 1 12519 | 12518 12519 | 99.98 | 175592 175592 | 7.13 7.13 | 7180000001443 7180000001443 [END]
[S1] [E1] | [S2] [E2] | [LEN 1] [LEN 2] | [% IDY] | [LEN R] [LEN Q] | [COV R] [COV Q] | [TAGS] =============================================================================================================================== 4790727 4911492 | 120764 1 | 120766 120764 | 99.98 | 6397126 175592 | 1.89 68.78 | gi|28867243|ref|NC_004578.1| 7180000001443 4898971 4955870 | 175592 118697 | 56900 56896 | 99.98 | 6397126 175592 | 0.89 32.40 | gi|28867243|ref|NC_004578.1| 7180000001443
Sanger AMOScmp (Chromosome+3 plasmids ref)
Reference=complete genome(chromosome+3 plasmids) use "circular contig" in Celera 3.11 assembly /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1012_AMOSCmp-relaxed-3plasmids 38 contigs: 15 for main chromosome, 1 for longer plasmid, 21 for shorter plasmid, 1 for "circular contig" The missoriented read pile corresponding to the chromosome (4. AMOSCmp of Sanger reads) has dissapeared AA ready for submission: /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1012_AMOSCmp-relaxed-3plasmids/AA/umd-20071030-141700.tar.gz
Solexa AMOScmp
1. align all reads (Solexa) to the reference using nucmer. I initially used minmatch=20, mincluster=20 (-c 20 -l 20)
6340136 reads 5641782 (88.98%) aligned by nucmer -c 20 -l 20 3453618 (54.47%) aligned by nucmer -c 32 -l 20 2707005 (42.69%) aligned by nucmer -c 32 -l 32
Solexa assemblied for different read coverages
Location
/fs/szasmg2/Bacteria/Pseudomonas_syringae/Assembly/Solexa/sample/ Several AMOScmp assemblies, using 10%,20%, ... 100%, of the P. syringae Solexa reads. These would correspond to 3X,6X ... 30X, coverage The read sampling was done randomly. One sample set for each coverage. all contigs desc #elem min max mean stdev sum 10 43136 32 7712 135.11 140.61 5828148 20 11243 32 20190 570.01 686.5 6408705 30 2972 32 27962 2185.32 2804.56 6494784 40 1058 32 63125 6152.98 7871.7 6509855 50 455 32 163430 14319.01 19663.15 6515153 60 267 32 328882 24406.61 46172.62 6516567 70 166 32 671064 39260.9 84200.42 6517311 80 143 32 906652 45577.16 111875.19 6517535 90 117 32 1433643 55708.4 164246.61 6517883 100 106 32 2067205 61489.83 230284.47 6517923 chromo contigs desc #elem min max mean stdev sum 10 42845 32 1845 133.32 118.36 5712348 20 11124 32 9650 565.41 625.32 6289649 30 2876 32 26076 2216.64 2714.92 6375063 40 965 32 63125 6621.71 7893.19 6389957 50 362 32 163430 17665.19 20565.31 6394800 60 167 32 328882 38299.32 53660.75 6395987 70 75 257 671064 85287.52 108858.19 6396564 80 49 940 906652 130546.42 160470.1 6396775 90 25 42603 1433643 255877.72 277650.54 6396943 100 18 42603 2067205 355387.77 465907.88 6396980 all gaps desc #elem min max mean stdev sum 10 43137 1 3874 16.46 38.01 710112 20 11242 1 3919 11.52 64.43 129555 30 2971 1 3418 14.63 114.29 43476 40 1056 1 3873 26.89 196.7 28405 50 454 1 3415 50.89 291.04 23107 60 265 1 3870 81.86 380.9 21693 70 165 1 3868 126.96 486.88 20949 80 141 1 3414 146.98 461.06 20725 90 115 1 3418 177.19 520.11 20377 100 104 1 3278 195.54 511.06 20337 chromo gaps desc #elem min max mean stdev sum 10 42846 1 240 15.98 16.33 684778 20 11125 1 146 9.66 9.72 107477 30 2876 1 76 7.67 7.73 22063 40 965 1 58 7.42 7.8 7169 50 362 1 48 6.42 7.08 2326 60 167 1 58 6.82 7.63 1139 70 76 1 55 7.39 7.9 562 80 49 1 55 7.16 10.08 351 90 25 1 45 7.31 10.12 183 100 18 1 55 8.11 13.62 146
CBCB (new)
Alignment based trimming
!!! Reduced the duplications significantly
Solution:
1. align all reads (Solexa) to the reference using nucmer. I initially used minmatch=20, mincluster=20 (-c 20 -l 20)
6340136 reads 5641782 (88.98%) aligned by nucmer -c 20 -l 20 3453618 (54.47%) aligned by nucmer -c 32 -l 20 2707005 (42.69%) aligned by nucmer -c 32 -l 32