Bumblebee: Difference between revisions
Jump to navigation
Jump to search
| Line 62: | Line 62: | ||
* Location: | * Location: | ||
/fs/szattic-asmg4/Bees/Bombus_impatiens | /fs/szattic-asmg4/Bees/Bombus_impatiens | ||
== D Kelly's trimming == | |||
438088072 total reads | |||
109166398 reads were thrown away | |||
148886138 reads were corrected and/or trimmed (to a min length of 30 bp) | |||
= Assembly = | = Assembly = | ||
Revision as of 21:24, 8 March 2010
Data
- ~ 500B genome
Traces
- 7 pairs of data files (paired ends) : lanes 1..3,5..8 (lane 4 wasn't used)
Lane Insert ReadLen #Reads Coverage Comments 1 3K(2..6,avg 4K) 124 34,944,099 14X 2 8K(7..9,avg 8K) 124 32,540,640 13X 3 500(450..600) 124 34,745,750 # gDNA 5 500 34,601,239 6 500 34,553,857 7 500 34,682,612 8 500 12,975,839
- Adaptors
>circularizarion CGTAATAACTTCGTATAGCATACATTATACGAAGTTATACGA >circularizarion.revcomp TCGTATAACTTCGTATAATGTATGCTATACGAAGTTATTACG >5 GATCGGAAGAGCGGTTCAGCAGGAATGCCGAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCG >3 CGGCATTCCTGCTGAACCGAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT
Tasks to figure out
- Erroneous reads/bases, which we need to correct or discard
- GC bias, so we can compute a-stats properly
- Redundancy in the long paired ends, which are lane 1 and lane 2.
- Used the 454 protocol to circularize the DNA for sequencing with the Illumina instrument.
- Some reads will begin in the circularization adaptor and thus will have only one usable read
- Some reads have a few bases of DNA sequence and hit the circularization adaptor right away
- Most reads will have at least 36bp from each end before hitting the adaptor.
- Many reads will not have any adaptor to trim (>125bp of DNA sequence at both ends of the adaptor)
Trimming
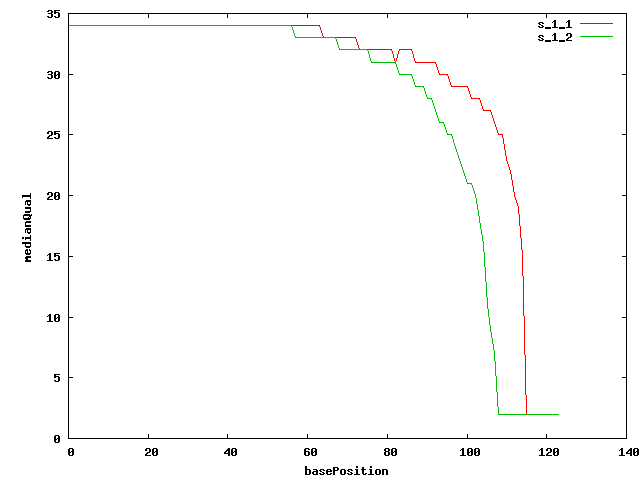
- Quality plots
- Keep only the first 100bp (last 24 bp are anyway low qual) otherwise gatekeeper "Seg fault"
- Adaptor trimming:
- Split data set in 1M read subsets
- Quality trimming
{kind=link}
cat s_1_*_sequence.*.txt | ~/bin/fastq2clb.pl > s_1_sequence.clb
- Vector trimming: Align all subsets to adaptors
nucmer -l 8 -c 16 -b 8 -g 8 adaptors.seq s_1_1_sequence.00.seq -p s_1_1_sequence.00 delta-filter -l 16 -q s_1_1_sequence.00.delta > s_1_1_sequence.00.filter-q.delta ... cat s_1_*_sequence.*.filter-q.delta | ~/bin/delta2clr53.pl -5 5,3 -minLen 64 > s_1_sequence.clv
- Stats
. elem <=64 >64 min q1 q2 q3 max mean n50 sum orig 69888198 0 69888198 124 124 124 124 124 124 124 8666136552 clq 69888198 7724022 62164176 0 89 111 124 124 96.76 117 6762346722 clv 69888198 18607136 51281062 0 0 124 124 124 86.96 124 6077231064 clr 69888198 24677952 45210246 0 0 88 115 124 67.31 113 4704368689
- Location:
/fs/szattic-asmg4/Bees/Bombus_impatiens
D Kelly's trimming
438088072 total reads 109166398 reads were thrown away 148886138 reads were corrected and/or trimmed (to a min length of 30 bp)
Assembly
- Trimming
No OBT adaptors in the seqs
- Kmers
meryl -Dh -s 0-mercounts/asm-C-ms22-cm1 >! 22mers.hist Found 3136399464 mers. Found 379123530 distinct mers. Found 201257394 unique mers. Largest mercount is 12006651; 90 mers are too big for histogram.
countKmers.pl most frequent 42mer : CGTAATAACTTCGTATAGCATACATTATACGAAGTTATACGA ~ 20% of the seqs : circularization adapter
- Overlapper
#overlaps/read reads 0count min q1 q2 q3 max mean n50 sum 62164168 21589472 0 0 4 12 324 11.42 38 709902310
- Unitigger : max utg len=852bp
- Consensus after unitigger : 3 out of 129 jobs failed
- Location
/fs/szdevel/dpuiu/SourceForge/wgs-assembler.030210/Linux-amd64/bin/runCA