Pseudomonas aeruginosa
Strain PA-b1
Data
CBCB
Reads:
8627900 33 bp Solexa reads
Coverage: ~ 43.55X
File location:
/fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/
Files:
s_1_sequence.txt # contains seq & qual; seq names: HWI% ; all reads are 33 bp s_1_0001_prb.txt # 27679 lines of 4*36 values in the -40..40 range s_1_tag.txt # unique seq count in s_1_sequence.txt s_7_sequence.txt # contains seq & qual; seq names HWU% ; all reads are 33 bp s_7_0300_prb.txt s_7_tag.txt wc -l s_1_sequence.txt s_7_sequence.txt 4105993 s_1_sequence.txt 4521907 s_7_sequence.txt 8627900 total
#all reads that contain at least 1 ambiguity grep -c N s_*seq s_1_sequence.seq:37933 s_7_sequence.seq:69669
#all reads that contain at least 2 ambiguities cat s_1_sequence.seq | perl -ane 'print $_ if(/N.+N/);' | wc -l : 18771 cat s_7_sequence.seq | perl -ane 'print $_ if(/N.+N/);' | wc -l : 59589 ...
s_1 reads have significantly fewer N's than s_7 reads !!! s_1 reads align better to ref than s_2 reads !!! Conflicts with avg qualities: 28.26 for s_1, 31.05 for s_2 !!! Base qualities: . #elem #elem0 #elem<0 min mean max sum s_1 135497769 1863585 3231899 -5 28.2624918053079 40 3829504586 s_7 149222931 1357384 3113806 -5 31.0508663175903 40 4633501282
NCBI
Same Strains
Ranging from 6.2M to 6.9M
PA14 CP000438 6,537,648 bp, 66.29 %GC : 5,892 genes most similar to PAb1 PAO1 AE004091 6,264,404 bp, 66.56 %GC : 5,568 genes rearrangement vs PA14 PACS2 AAQW01000001.1 6,492,423 bp, 66.33% GC : 5,676 genes rearrangement vs PA14 PA2192 . 6,905,121 bp, 65.99% GC : 6,157 genes rearrangement vs PA14 PAC3719 . 6,222,097 bp, 66.30% GC : 5,650 genes no rearrangement vs PA14 PA7 CP000744 6,588,339 bp : 6,286 genes no rearrangement vs PA14
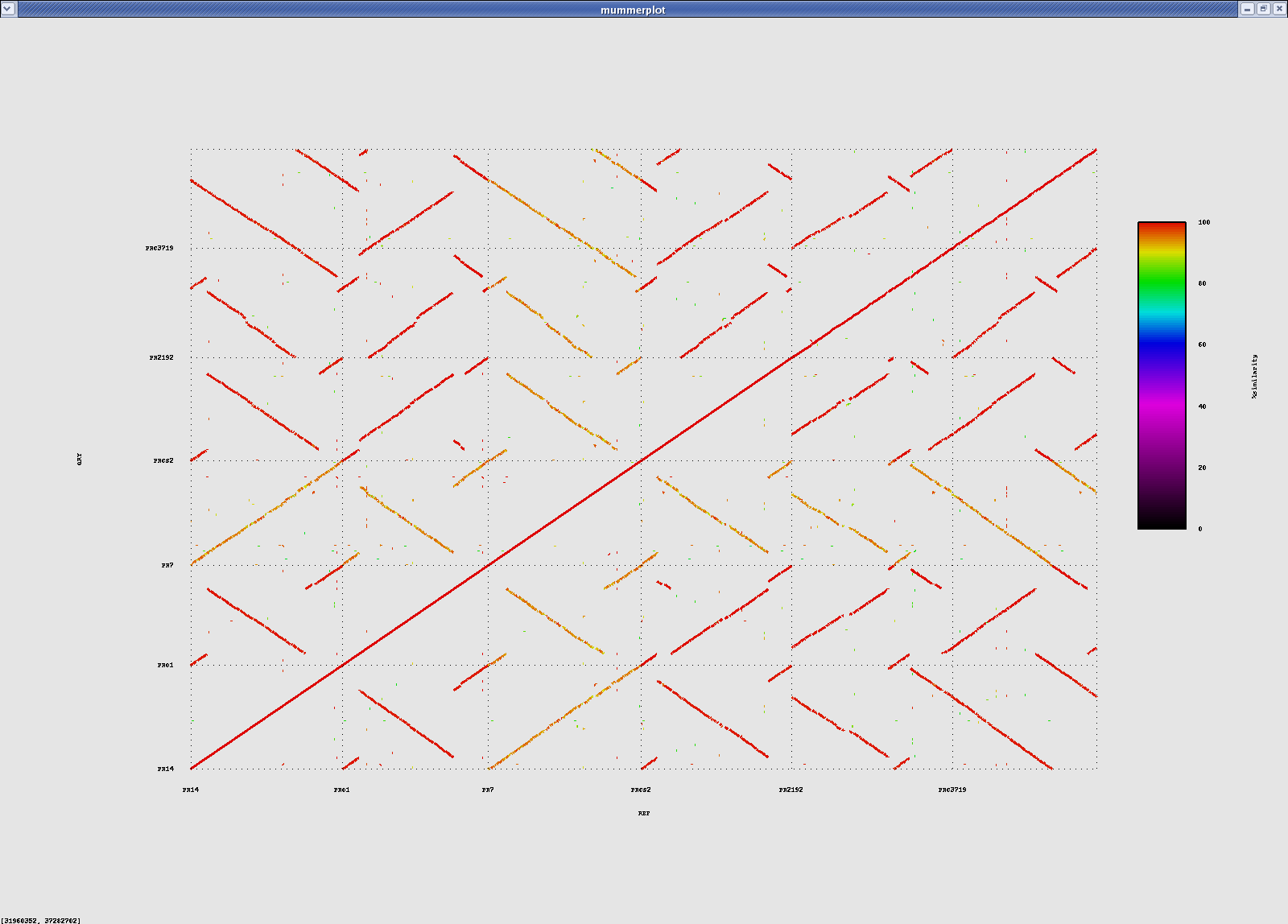
All vs all nucmer alignments:
#ref-qry count sum avg%id PA14-PAO1 404 6296632 98.66 PA14-PACS2 462 6303773 98.56 PA14-PA2192 532 6248297 98.47 PA14-PAC3719 604 6129432 98.44 PA14-PA7 822 5899920 93.72
Incomplete strains details(Broad):
Contig length stats: desc #contigs min max mean stdev sum C3719 124 2079 242903 49572 53770 6146998 2192 82 2087 398738 83246 88681 682625
5.7Kbp repeat: contains 16S rRNA gene; identical in all the strains
Coordinates: PA14: 732540 738302 + 4951956 4957714 - 5535975 5541728 - 6312434 6318199 -
PAO1: 721550 727325 + 4788516 4794273 - 5264042 5269801 - 6039515 6045289 -
PA7: 806558 812299 + 4982874 4988600 - 5566182 5571924 - 6353418 6359148 -
Pa's vs Pa's (nucmer)
{kind=link}
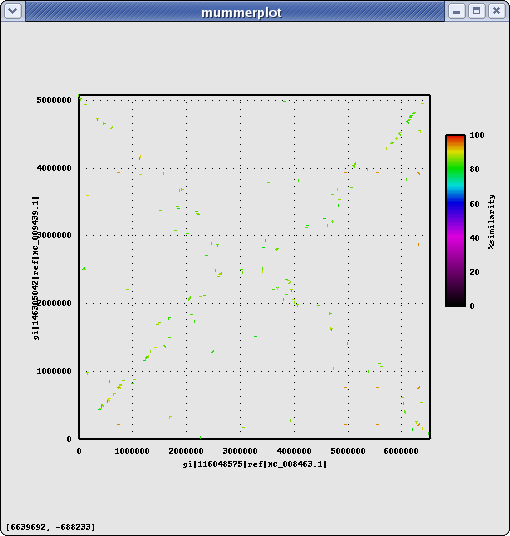
Same Genus, same Group, different Species: Pseudomonas mendocina ymp (Pm)
Name Length %GC Info NC_009439.1 5072807 64.68 PA14 vs Pm (nucmer) #elem min max mean median n50 sum avg%id Alignments(1con-1con : nucmer) 1281 75 19618 1256 1011 1548 1609296 85.26 Alignments(genes-genes: nucmer) ... 1326 PA14 genes align to 1306 Pm genes Alignments(genes-genes: promer) ... 2924 PA14 genes align to 2957 Pm genes
{kind=link}
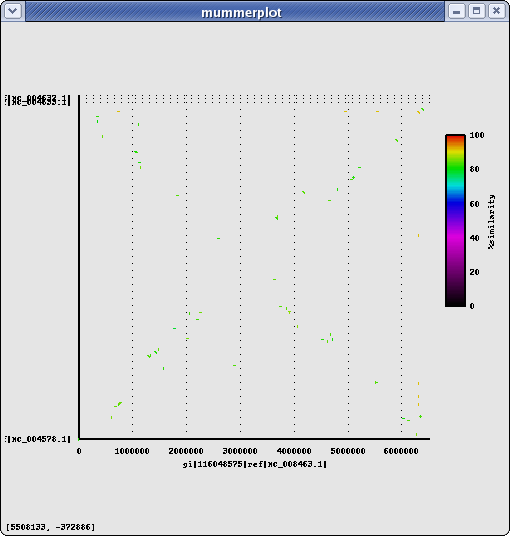
Same Genus, different Species: Pseudomonas syringae pv. tomato str. DC3000 (Ps) : chomosome+2plasmids
Name Length %GC Info NC_004578.1 6,397,126 58.40 chromosome NC_004633.1 73,661 55.15 plasmid pDC3000A NC_004632.1 67,473 56.17 plasmid pDC3000B total 6,538,260 PA14 vs Ps (nucmer) #elem min max mean median n50 sum avg%id Alignments(1con-1con : nucmer) 499 75 9435 1296 1161 1619 646614 84.79 Alignments(genes-genes: nucmer) 438 161 4446 1090 996 1281 477229 83.38 425 PA14 genes align to 418 Ps genes Alignments(genes-genes: promer) 6193 114 13026 1159 1011 1359 7180527 67.36 2556 PA14 genes align to 2644 Ps genes
{kind=link}
Publications
- Dynamics of Pseudomonas aeruginosa genome evolution PNAS Feb 2008
* 5,021 genes that are conserved across all five genomes analyzed, with at least 70% sequence identity (core genome) * Of these, >90% of the genes share at least 98% sequence identity * The genome for each strain carries a relatively modest number of unique sequences, 10% or less of the number in the core genome, ranging from 79 in C3719, to 507 in PA2192. * Less than 100 genes are unique to pairs of genomes, whereas the number decreases significantly when comparison is carried out among sets of three or four genomes. * The overall architecture of the pangenome of P. aeruginosa an be represented as a circular chromosome with dispersed polymorphic strain-specific segments, flanked by conserved genes referred to as anchors * These strain-specific segments are regions of genomic plasticity (RGP) and include any region of at least four contiguous ORFs that are missing in at least one of the genomes analyzed. * The number of RGPs in individual genomes varies from 27 to 37. A total of 52 RGPs were identified in the five genomes analyzed
Read trimming
No trimming
If no trimming is done and the default AMOScmp pipeline is used (ALIGNWIGGLE=15), the contigs end up containing reference duplications Make-consensus will also take a very long time to run
Duplication causes:
- Only the reference nucmer alignment coordinates are stored in the bank and used by casm-layout and make-consensus; the read alignment coordinates are not stored, just the read clear ranges (the ones given in the .afg file); read positions in the assembly layout are approximate
- reads can be shifted from their original alignment position by make-consensus by up to 2^3*15=120 bp (constants ALIGN_WIGGLE=15; MAX_ALIGN_ATTEMPTS=3)
- The Pseudomonas_aeruginosa reference contains multiple 2 copy 5-10 bp kmers, adjacent within a few dozen bp of each other; reads starting with these kmers align in 2 ways to the reference; if the reads contain errors or SNP's, the 2nd (shorter) alignment to the greedyly built consensus is chosen over the correct one and a duplication is introduced
Quality trimming
Art's script: qual-trim.awk
Min_Qual=15
. #elem #elem0 #elem<0 min median max sum mean stdev n50 qualTrim 8627900 1266 0 0 23 33 193999902 22.49 7.43 26
!!! Avg clr drops from 33 to 22 bp !!! Duplications still exist though at a lower rate
Alignment based trimming (nucmer)
PA14 is the most similar strain: more PAb1 reads aligned to PA14 than to PAO1 or PA7
grep -c "^>" PA14-Pa.l-17_c-17.delta PAO1-Pa.l-17_c-17.delta PA7-Pa.l-17_c-17.delta PA14-Pa.l-17_c-17.delta:7261629 PAO1-Pa.l-17_c-17.delta:7036243 PA7-Pa.l-17_c-17.delta:5393144
Read mapping
PA14 reference
Commands:
blat -noHead -t=dna -q=dna -tileSize=10 -stepSize=3 Pa.1con Pa.seq Pa.blat nucmer.pl -l 20,16,12 -c 20,16,14 Pa.1con Pa.seq -p Pa.nucmer rmap -m 3 -w 33 -c Pa.1con Pa.seq -o Pa.rmap soap -v 5 -d Pa.1con -a Pa.seq -o Pa.soap
Results:
runTime hits(all) hits(>=30bp) blat: 1038.28u 3.696s 17:49.00 97.4% 7004925 6353293 nucmer: . 7553734 6832230 # 3 itterations were run for nucmer -l 20,16,12 -c 20,16,14; delta-filter -l 20 rmap: 7318.77u 5.770s 2:02:11.29 99.9% 6958043 6958043 soap: 685.208u 3.971s 12:45.68 90.0% 7310025 7310025 # fastest, most hits all: . 7638420 7201727
Soap seems the "best" read mapping program at current moment.
#number of hits/read
cat Pa.soap.5 | awk '{print $4}' | count.pl
# total
1 7185054 # most reads align in only one place
2 83418
3 8706
4 32277
5 291
...
Total 7310025
#number of misamtches
awk '{print $10}' Pa.soap.5 | count.pl
# total
0 3708583 # that many reads align exactly to the reference
1 2025346
2 908998
3 395002
4 181326
5 90770
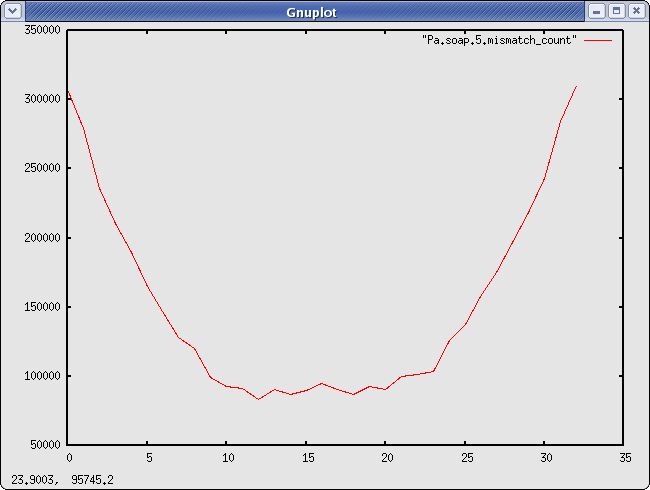
#positions of the mismatches inside a read Pa.soap.5.mismatch_count There are ~3 times more mismatches at the beginning/end of the read than in the middle
{kind=link}
soap -v 5 -s 12 -g 3 -d Pa.1con -a Pa.seq -o Pa.soap
Pm reference
Reads aligned: 1182734
cat Pm.soap | awk '{print $10}' | count.pl | sort -n
#number of misamatches
0 48396
1 117192
2 207369
3 267275
4 279223
5 263279
Total 1182734
Ps reference
Reads aligned: 632459
cat Pm.soap | awk '{print $10}' | count.pl | sort -n
#number of misamatches
0 16442
1 36440
2 81147
3 136472
4 176779
5 185179
Sequence Assemblies
Comparative
PA14 ref assembly
Location: 2008_0109_AMOSCmp-PA14-relaxed-17-nucmer-redo2 Command: AMOScmp Pa -D MINCLUSTER=17 -D MINMATCH=17 -D MINOVL=5 -D MAJORITY=50 -D ALIGNWIGGLE=2
- a align all reads to the reference using nucmer. I initially used minmatch=17, mincluster=17 (nucmer -c 17 -l 17)
8.62M reads, 4.10M HWI 5.32M HWU 8.30M (...) total reads align to the reference (nucmer -c 14 -l 14) 7.59M (...) total reads align to the reference (nucmer -c 16 -l 16) 7.25M (...) total reads align to the reference (nucmer -c 16 -l 16; delta-filter -l 20) 7.26M (84.16%, 83.50% HWI, 84.76% HWU) total reads align to the reference (nucmer -c 17 -l 17) 6.54M (75.82%, 74.49% HWI, 77.02% HWU) total reads align to the reference (nucmer -c 20 -l 20) 5.17M (...) total reads align to the reference (nucmer -c 28 -l 14) 5.32M (73.27%) reads aligned on their full length (as opposed to 0.94M 33bp quality untrimmed reads) (nucmer -c 33 -l 17) 3.69M (42.79%, 38.24% HWI, 46.92% HWU) align exactly (33 bp, 100%id)
7.00M align using blat -t=dna -q=dna -stepSize=5 -tileSize=10 (20bp alignments, max 3 mismatches) 6.35M align using blat -t=dna -q=dna -stepSize=5 -tileSize=10 (30bp alignments filtered from above results) 7.11M align using blat -t=dna -q=dna -stepSize=3 -tileSize=10 (20bp alignments, 3 mismatches) 500K the unaligned ones were aligned by blat -t=dnax -q=dnax -stepSize=2 -tileSize=4 (20bp alignments, 6 mismatches; only 15% of the aligned reads are unique) => 7.61M aligned by blat/blatx
6.95M aligned by rmap (-m 3)
7.30M align by soap (-v 5 -s 12 -g 3) to PA14 chromosome 6.64M align by soap (-v 2 -s 12 -g 3) to PA14 chromosome 7.24M align by soap (-v 2 -s 12 -g 3 -c 42) to PA14 chromosome (iterative trimming) 6.72M align by soap (-v 1 -s 12 -g 3 -c 41) to PA14 chromosome (iterative trimming)
6.36M align by soap (-v 5 -s 12 -g 3) to PA14 genes
7.43M align by maq
- b align all unaligned reads to the reference using nucmer. minmatch=14, mincluster=14 (-c 14 -l 14)
- Combine 1a & 1b
- trim reads according to their nucmer alignment coordinates; don't trim the ones adjacent to zero cvg regions
- assemble them using the AMOScmp pipeline(ALIGN_WIGGLE=2 instead of 15)
Contigs stats: desc #elem min max mean stdev sum singletons all 2053 17 170485 3011.84 11917.53 6183320 1127399 200 428 203 170485 14262.09 22852.74 6104175 10K 157 10240 170485 35468.89 26531.33 5568616 Data accuracy Get all assembled bases with coverage>=20 count_bases=5926977 sum_bases=235619001 sum_errors=2455670
sum_errors/sum_bases=2455670/235619001=0.01042=1.042% error
SNP's in the 157 >10K contigs: T<->C & A<-G> the most common
PA14 PAb1 count T C 6153 C T 6100 G A 6075 A G 6021 G C 1357 C G 1309 G T 859 C A 846 T G 840 A C 836 A T 338 T A 308 G . 241 C . 196 . C 182 . G 152 A . 101 T . 88 . T 74 . A 70 Total 32146
Problems:
- Some of the short ctgs are not "real"
- Some misassemblies due to loose assembly params (MINOVL=5 MAJORITY=50 ): Example: pos 6700
PA14 ref assembly (2) *
run 3 nucmer iterations
Parameters:
MINCLUSTER=20,16,14 # nucmer -c MINMATCH= 20,16,12 # nucmer -l MINLEN=20 # delta-filter -l MINOVL=10 # casm-layout -o ; previously 5, allowed for missasemblies MAJORITY=70 # casm-layout -m ; previously 50 to reduce the number of negative gaps
Read CLR's:
33 5510573 32 471289 31 456959 30 412738 29 136423 28 120148 27 104213 26 60338 25 54828 24 44854 23 38574 # smallest value !!! 22 43302 21 49923 20 49572 Total 7553734
Contig stats:
desc #elem min max mean median n50 sum singl breaks all 1843 20 127984 3350 63 30995 6174765 1107200 9 200bp+ 542 200 127984 11245 3263 31106 6094904
Alignments of PA14 genes to the contigs
show-coords ctg-genes.filter-1.delta | awk '{print $20}' | count.pl
# total
[CONTAINS] 5318
[END] 198
[BEGIN] 191
[CONTAINED] 190
[PARTIAL] 88
[IDENTITY] 3
Location:
/fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0424_AMOSCmp-PA14
PA14 ref assembly (used blat instead of nucmer for read alignment)
run 2 blat iteration:
blat -t=dna -q=dna -stepSize=3 -tileSize=10 Pa.1con Pa.seq blat -t=dnax -q=dnax -stepSize=2 -tileSize=4 Pa.1con Pa.singletons.seq
Contig stats:
#ctgs min max mean median n50 sum singletons all 1372 20 176323 4517 64 47563 6197771 1049302 200bp+ 370 201 176323 16584 3929 47563 6136095 10Kbp+ 146 10097 176323 39176 30457 51554 5719752
Location:
/fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0613_AMOSCmp-PA14-blat
!!! Best AMOScmp assembly so far : * fewer contig than when using nucmer; * largest contig * largest contig sum
PA14 ref assembly (used soap instead of nucmer for read alignment)
- soap parameters: -v 5 -g 3 -s 12
Contig stats:
#elem min max mean median n50 sum
all 1228 33 132817 5022 106 33779 6167051 !!! smaller assembly 6.16M vs 6.19M with blat
200bp+ 478 200 132817 12789 3824 33988 6113109
10Kbp+ 303 1000 132817 19927 12470 34129 6037852
Location: /fs/szasmg3/dpuiu/Pseudomonas_aeruginosa/Assembly/2008_0625_AMOSCmp-PA14-soap/
- soap parameters: -v 2 -g 3 -s 12 -c 42 :
- iterative trimming of 2 bp from read ends
Contig stats:
#elem min max mean median n50 sum
all 1932 33 79489 3190 153 16122 6162793
200bp+ 874 200 79489 6960 3195 16145 6082694
10Kbp+ 202 10048 79489 20745 17422 21640 4190588
PAO1 ref assembly
Same method except that 1.b was not used
Contigs stats: desc #elem min max mean stdev sum all 2797 17 75626 2161.19 5812.2 6044851 200 865 200 75626 6893.96 8766.63 5963278 10K 204 10016 75626 19016.22 10368 3879309 Singletons: 1592525
Location:
/fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0124_AMOSCmp-PAO1-relaxed-17-nucmer-redo2
PAO1 ref assembly (2)
Parameters:
MINCLUSTER=16 # nucmer -l MINMATCH=16 # nucmer -c MINLEN=20 # delta-filter -l MINOVL=10 # casm-layout -o ; previously 5, allowed for missasemblies MAJORITY=70 # casm-layout -m ; previously 50 to reduce the number of negative gaps
Contig stats:
desc #elem min max mean stdev sum all 1961 20 46414 3065 5251 6010800 200bp+ 1166 203 46414 5108 6007 5955960 1758777 singletons
Location:
/fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0424_AMOSCmp-PAO1
PA14 & PAO1 merge
Use minimus to merge all contigs in the PA14 & PAO1 reference assemblies
Filter contigs that contain at least 2 adjacent PA14 merged by PAO1
desc #elem min max mean stdev sum all 1850 17 236472 3400.31 16863.79 6290586 200 306 204 236472 20318.3 37147.91 6217401 10K 113 10520 236472 52647.45 45602.76 5949162 Singletons: 1066226
De novo assembly of singletons using SSAKE Input: 1066226 singletons
Ssake version 3.0 run with default parameters: -m Minimum number of reads needed to call a base during overhang consensus build up (default -m 16) -o Minimum number of reads needed to call a base during an extension (default -o 2) -r Minimum base ratio used to accept a overhang consensus base (default -r 0.6)
Contigs stats: desc #elem min max mean stdev sum all 19795 34 2866 67.78 102.73 1341825 200 879 200 2866 416.72 308.92 366304
200 bp contigs stats desc. #elem min mean max sum contig_len 879 200 416.72 2866 366304 contig_reads 879 52 414.21 3157 364091 contig_cvg 879 7.91 31.58 146.52 27764.51 Singletons: 702007
Find contigs overlapping 12 bp or more and merge them using EMBOSS merger program: desc #elem min max mean stdev sum original 879 200 2866 416.72 308.92 366304 new 670 200 4826 539.32 579.26 361350
Pa-b1 AMOScmp reference assembly
Contig stats: desc #elem min max mean stdev sum input(reference|final) 770 200 512638 8484.21 37210.58 6532844 output(AMOScmp contigs) 936 21 260827 6988.98 22288.49 6541689 Singletons: 1257963 Snp's: 1941
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0215_AMOScmp-PAb1
Use ALL strains as reference
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0228_AMOSCmp-ALL
Steps:
* Identify related strains (completed & draft assemblies): PA14, PAO1, PA7, PACS2, PA2192, PAC3719 * Download & merge related strains into a multi-FASTA file * Align PAb1 reads to multi-FASTA file using nucmer (Ex: -l 17 -c 17) * Count the number of PAb1 reads aligned to each strain
strain #reads_aligned
======================
PA14 7261629
PA2192 7193960
PACS2 7089455
PAO1 7036243
PAC3719 6928744
PA7 5393144
======================
* Sort the strains in descending order according to the number of reads aligned to each one * The strain with most hits is PA14 => closest relative
* Assuming the order is PA14, PAO1, PA7, PACS2, PA2192, PAC3719 ... * Align strains to one other; identify unique regions in each genome (regions which are not present in the previously considered ones) Unique regions length summary:
Table 1: reference #regions min max mean stdev sum ======================================================================================= PA14 1 6537648 6537648 6537648 0 6537648 PAO1-PA14 115 1 19263 2038.58 3843.93 234437 PA7-PA14 511 1 46020 1876.96 4872.51 959129 PACS2-PA14 147 1 90591 3054.19 8766.3 448967 PA2192-PA14 231 1 92553 3746.3 11475 865396 PAC3719-PA14 272 1 14058 1164.78 2089.85 316822 ======================================================================================= ALL 1277 9362399
Table 2: reference #regions min max mean stdev sum ======================================================================================= PA14 1 6537648 6537648 6537648 0 6537648 PAO1-PA14 115 1 19263 2038.58 3843.93 234437 PA7-PAO1-PA14 487 1 46020 1830.82 4910.86 891614 PACS2-PA7-PAO1-PA14 71 2 79031 4112.94 10618.2 292019 PA2192-PACS2-PA7-PAO1-PA14 181 1 92066 3439.9 9500.07 622622 #some N's maybe? PAC3719-PA2192-PACS2-PA7-PAO1-PA14 173 1 11425 998.16 1888.94 172682 #some N's maybe? ======================================================================================= ALL 1028 2 6537648 8513.66 203929 8752049
Table 3: reference #regions min max mean stdev sum ======================================================================================= PA14 1 6537648 6537648 6537648 0 6537648 PA14-PAO1 123 1 43690 4009.28 7903.16 493142 PA14-PA7 516 1 31464 1768.16 3534.88 912375 PA14-PACS2 146 2 43690 3259.15 6129.12 475836 PA14-PA2192 222 1 34854 2189.76 4338.6 486127 PA14-PAC3719 277 1 43690 2206.38 5214.55 611170 =======================================================================================
* Extract unique regions(Table 2) and add them to the most similar genome
* Assemble using all these regions as reference => 10,201 contigs; 1,052,178 singletons
reference #ctgs min max mean stdev sum ======================================================================================= PA14 2056 17 57476 2992.27 7078.25 6152122 PAO1 877 17 8159 95.16 482.74 83456 PA7 4723 17 6822 41.7 128.87 196996 PACS2 914 17 3346 66.96 148.47 61207 PA2192 1528 17 49826 134.88 1734.19 206109 PAC3719 103 17 693 42.11 85.89 4338 ======================================================================================= ALL 10201 17 57476 657.21 3457.1 6704228 * Compare ALL assembly contigs to PA14 ref assembly :
reference #ctgs min max mean stdev sum #singletons ======================================================================================================== ALL 10201 17 57476 657.21 3457.1 6704228 1052178 ALL(200bp+) 1020 200 57476 6235.17 9220.76 6359881 ALL(PA14) 2056 17 57476 2992.27 7078.25 6152122 PA14 1984 17 82196 3103.21 8274.53 6156788 1375004 ======================================================================================================== ALL-PA14 8217 ...... 547440 # 0.5M extra bp ALL(PA14)-PA14 72 ...... -4666 # 4666 bp which were originally mapped to PA14 are now mapped to other strains
* Compare ALL assembly contigs to ssake 200bp+ contigs :
ssake 200bp+ contigs:
strain #ctgs min max mean stdev sum
PAb1 879 200 2866 416.72 308.92 366304
ALL vs ssake alignments:
desc #elem min max mean stdev sum
ALL 265 23 49826 912.9 4187.17 241919
ssake 306 200 2358 379.85 257.04 116235
ALL vs ssake CONTAINED alignments:
desc #elem min max mean stdev sum
ALL 164 23 49826 1264.71 5268.16 207413
ssake 260 200 2358 368.09 259.02 95704
~300 out of 879 ssake contigs were assembled using this method. The ALL assembly contigs ssake contigs align to are longer than the ssake contigs!
* Compare ALL assembly contigs to final de novo contigs :
id len #de novo ctgs it CONTAINS
======================================================================
1 2361_PA2192_2249428_2341494 49826 52
2 2360_PA2192_2249428_2341494 42258 49
3 2364_PA2192_2346558_2361947 15388 17
4 10190_PAO1_747565_761293 8159 7
5 2429_PA2192_2405271_2412668 6103 7
6 3259_PA2192_6656583_6693732 2594 6
7 2367_PA2192_2374235_2379148 4914 5
8 6917_PA7_4563267_4570077 6822 5
9 2958_PA2192_4496915_4500004 3090 4
10 2375_PA2192_2383706_2386485 2789 3
11 2366_PA2192_2366515_2369415 2901 2
12 6919_PA7_4579629_4582081 1085 2
13 5364_PA7_3138605_3164438 1931 2
14 8663_PACS2_3494057_3573088 449 2
15 9322_PACS2_982458_983976 1519 2
16 2368_PA2192_2379432_2380661 1230 2
17 2716_PA2192_3268239_3292117 567 2
18 9587_PAO1_2439014_2458239 3513 1
...
49 8384_PAC3719_5517906_5518496 591 1
======================================================================
Total 201
PA14 Sanger maq on all reads
Contig stats: min_len #contigs min max mean stdev sum 0 991 33 155551 6199.79 17445.05 6143996 !!!Smaller assembly size than with AMOScmp 200 368 200 155551 16581.7 25475.92 6102067 10K 149 10048 155551 38164.35 28513.51 5686489
1197385 singletons; SNP's in the contigs: T<->C & A<-G> the most common
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0206_PA14_maq
PA14 gene ref assembly
- Use the 5892 PA14 genes as reference
- Use "soap -v 5 -s 12 -g 3" for alignments
=> 5671 scaffolds (that many genes are shared by the 2 strains)
contig stats:
#elem min max mean median n50 sum all 6487 33 13070 854 752 1164 5541032 200bp+ 5666 200 13070 965 840 1174 5470492
Comparative-ABBA
ABBA
Steps
- Annotate draft assemby
- Find fragmented genes at ends of contigs. Example shown is of putative secretion protein from PAO1
- Fill in missing amino acid sequence using reference protein
- Merge contigs together with output sequence from ABBA
Pipeline
- Input reference amino acid sequence and database of short reads
- Search read database (tblastn)
- Create layout using alignment coordinates
- Validate good read coverage across entire sequence
- Make consensus from a multiple alignment
PA14:PO1
- 5892 genes in PA14 vs 5568 in PAO1
- 5244 genes in PA14 IDENTITY (99.03%) to 5239 genes in PAO1
- 5379 genes in PA14 IDENTITY|CONTAIN|BEGIN|END (99.003%) to 5367 genes in PAO1
- 5389 genes in PA14 align to 5372 genes in PAO1 (98.69%id)
- 501 genes in PA14 & 196 genes in PAO1 don't align to opposite strand genes
!!! The genes that align are very conserved
PA14 gene ref assembly
desc #elem min max mean stdev sum genes(nt) 5892 72 15639 992 752 5846175 # out of a total of 6537648bp genome => 89.42 coding genes(aa) 5892 23 5212 330 251 1942833
PAO1 gene ref assembly
desc #elem min max mean stdev sum genes(nt) 5568 72 16884 1006 751 5601468 # out of a total of 6264404bp genome => 89.41 coding genes(aa) 5568 23 5627 334 250 1861588
De-novo
SSAKE
version 3.0 with default parameters
Contigs stats: desc #elem min max mean stdev sum length 185030 34 5490 77.21 141.23 14287079 reads 185030 2 13352 29.52 127.65 5463405 Singletons: 3,164,495 (singletons file + ambiguous reads)
Run times:
18459.472u 31.849s 5:08:59.82 99.7% 0+0k 0+0io 0pf+0w
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0214_ssake/
Latest version is 3.2: run on the same dataset generated fewer contigs and more singletons; also took longer to run
Velvet *
Version 0.5.05 Ovl len=23
Commands:
host: sycamore (Opteron) velveth . 23 Pa.seq velvetg . -read_trkg yes
Run times:
velveth : 88.614u 22.865s 2:16.35 81.7% 0+0k 0+0io 0pf+0w velvetg : 439.939u 19.258s 8:19.77 91.8% 0+0k 0+0io 0pf+0w total : 528.553u 42.123s 10:36.12
Output files (used)
contigs.fa : contig FASTA file
Contigs stats:
#elem min max mean median n50 sum all 10684 45 16239 640 363 1184 6841458 200bp+ 7382 200 16239 877 586 1252 6474426 breaks 257 (-b 20) breaks(200+) 251 (-b 20) coming from 170 ctgs 1,273,164 singletons; 879,840 of them aligned by soap (-v5,-s12,-g3) to PA14
Distance between breakpoints:
#elem min max mean median n50 sum 251 0 314620 24946 3776 99406 6261404
Align velvet ctgs to PA14 ref (nucmer -c 40; delta-filter -q):
PA14 0cvg regions
#elem min max mean median n50 sum original 2235 1 31503 197 22 7529 439633 # 6098014 of PA14 covered delta-filter -q 2378 1 31676 220 24 6434 522246 # 6015401 of PA14 covered
rRNA repeat is collapsed; Contigs aligned to the 4 copy 5.7Kbp PA14 rRNA repeat:
[S1] [E1] | [S2] [E2] | [LEN 1] [LEN 2] | [% IDY] | [LEN R] [LEN Q] | [COV R] [COV Q] | [TAGS]
===============================================================================================================================
23 409 | 1 388 | 387 388 | 97.94 | 5763 388 | 6.72 100.00 | rRNA 856 [CONTAINS]
388 2132 | 1 1745 | 1745 1745 | 99.94 | 5763 1745 | 30.28 100.00 | rRNA 642 [CONTAINS]
2111 2279 | 169 1 | 169 169 | 100.00 | 5763 169 | 2.93 100.00 | rRNA 3433 [CONTAINS]
2258 5763 | 1 3506 | 3506 3506 | 99.89 | 5763 3506 | 60.84 100.00 | rRNA 260 [CONTAINS]
Question: Are there some velvet contigs contained in each other? Answer: Very few: 302 (out of 10K) ; avg %id is 86 and avg lenght is 106 bp; nucmer -c 20 has been used for alignments
Many contigs seem to overlap by ~ 20bp
Some contigs share end reads labeled as D(uplicates)
listReadPlacedStatus Pa.bnk | awk '{print $3}' | count.pl
# total
P 7322684
S 1273164
D 32052 # 33 of them belong to 3 contigs
Total 8627900
About ~4607 contig pairs could be merged:
grep D Pa.status.detailed | awk '{print $5,$6}' | sort -u | wc -l
4607
Most of the overlaps are shorter than 10bp; if "nucmer -c 20 -l 20" is run on the contig ends; only 627 of them are found
Example:
Contigs 1915 & 3551 share 15 Duplicate reads cat Pa.status | grep 1915 | grep 3551$ | wc -l 15 They align to adjacent pos in PA14 : 3743092 3745646 | 2555 1 | 2555 2555 | 99.49 | 6537648 2555 | 0.04 100.00 | 1 1915 [CONTAINS] 3745625 3746382 | 758 1 | 758 758 | 99.60 | 6537648 758 | 0.01 100.00 | 1 3551 [CONTAINS]
411870 out of 1273164 singletons (~33%) align to PA14 (nucmer -maxmatch -c 20) 304162 out of 1273164 singletons (~25%) are also singletons in the AMOScmp-PA14 assembly; probably the common ones are low quality sequences that just don't assemble
PA14 reference alignments: nucmer -maxmatch -c 40):
Aligned:
#elem min max mean stdev sum 9859 45 15342 642 785 6331357
Completely aligned:
#elem min max mean stdev sum 9681 45 11120 617 712 5969126
9208 are aligned 1 time 408 are aligned 2 times #repeats 39 are aligned 3 times #repeats 25 are aligned 4 times #repeats 1 is aligned 6 times #repeat
Partially aligned:
#elem min max mean stdev sum 178 78 15342 2035 2139 362231 120 are aligned once: Pa.1con-velvet.uniq.not_contained.filter-q.1.coords 58 are aligned more than once: Pa.1con-velvet.uniq.not_contained.filter-q.2.coords
All these contigs have AMOScmp assembly singletons aligned to them!!! These velvet contigs seem correct.
Example: There is a large insertion in PA14 at position 6700 PA-b1 reads vs PA14 ref find-query-breaks -B Pa.filter-q.delta => 3 reads end at base 6700 velvet ctgs vs PA14 ref: ... 6177 6700 | 1 524 | 524 524 | 99.43 | 6537648 875 | 0.01 59.89 | 1 900 # 431 out of 1054 seqs in this contigs are singletons in AMOScmp assembly 6694 8045 | 743 2094 | 1352 1352 | 99.63 | 6537648 2097 | 0.02 64.47 | 1 1970 # 895 out of 2354 seqs in this contigs are singletons in AMOScmp assembly; unaligned regions aligns at low %id to a Pseudomonas fluorescens hypothetical protein
Nucmer/Soap alignments of the reads adjacent to position 6700 Media:PA14.coord.6700 Media:PA14.soap.v5.s12.g3.6700 Media:PA14.soap.v2.s12.g3.6700
Other breaks: ... 66764 67073 | 1 310 | 310 310 | 100.00 | 6537648 1282 | 0.00 24.18 | 1 320 67181 67322 | 1136 1277 | 142 142 | 98.59 | 6537648 1282 | 0.00 11.08 | 1 320 ... 116429 117249 | 1 821 | 821 821 | 99.03 | 6537648 1457 | 0.01 56.35 | 1 1955 120490 120573 | 84 1 | 84 84 | 100.00 | 6537648 1693 | 0.00 4.96 | 1 1827 ... 234641 235980 | 3294 1955 | 1340 1340 | 99.40 | 6537648 3294 | 0.02 40.68 | 1 139 235981 237442 | 1466 5 | 1462 1462 | 99.32 | 6537648 3294 | 0.02 44.38 | 1 139 ... 286003 287915 | 1216 3128 | 1913 1913 | 99.22 | 6537648 3128 | 0.03 61.16 | 1 3068 ...
Not aligned:
#elem min max mean stdev sum 825 45 16239 618.3 1209.37 510101 # the longest velvet ctg does not align
Not CONTAINED:
#elem min max mean stdev sum 1002 45 16239 868.82 1518.56 870566
Velvet vs all Pseudomonas strains:
strain aligned CONTAINED partially-aligned PA14 9859 9681 178 # smallest number PA2192 9940 9607 333 PACS2 9688 9469 219 PAO1 9587 9402 185 PAC3719 9427 9057 370 PA7 8514 7651 863 all 10378 10243 135
not aligned stats: #elem min max mean median n50 sum 306 45 9681 549 119 2310 168094
strain(s) CONTAINED PA14 9681 PA14|PA2192 10081 PA14|PA2192|PACS2 10186 PA14|PA2192|PACS2|PAO1 10198 PA14|PA2192|PACS2|PAO1|PAC3719 10210 PA14|PA2192|PACS2|PAO1|PAC3719|PA7 10243
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0311_velvet-24
Velvet version 0.5.07 : does not "seem" to be an improvement over Version 0.5.05 Ovl len=23
desc #elem min max mean stdev sum contig 11230 45 16239 611.64 814.09 6868784 contig(200bp+) 7353 200 16239 878.61 897.06 6460467
Some of the 306 contigs should probably assemble together:
- Assembled with minimus2 REFCOUNT=0 OVL=20 => 54 ctgs that contain 132 reads & 174 singlets
- => reduced the number from 306 to 54+174= 228
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0311_velvet-24/no_hits
Edena
Version 2.1.1 , default parameters
Contigs stats:
#elem min max mean median n50 sum
all 11180 100 11300 552 356 837 6175460
200bp+ 8316 200 11300 693 488 902 5759209
breaks(PA14) 245 (-b 20) # 78 of them also present in velvet
breaks(velvet) 210 (-b 20)
3955865 singletons
Contig don't seem to overlap much
Run times:
ovl step: 1655.561u 7.560s 27:54.62 99.3% 0+0k 0+0io 0pf+0w assembly step: 56.141u 3.791s 1:03.82 93.9% 0+0k 0+0io 0pf+0w total: 1711.702u 11.351s 28:58.44
A significant number of edena contigs CONTAIN one(several) velvet contigs:
$ show-coords -I 94 velvet-edena.delta | egrep 'CONTAINED|BEGIN|END' | awk '{print $19}' | count.pl -m 1 | wc -l # 2269
$ show-coords -I 94 velvet-edena.delta | egrep 'CONTAINED|BEGIN|END' | awk '{print $19}' | count.pl -m 2 | wc -l # 1455
Align edena ctgs to PA14 ref (nucmer -c 40; delta-filter -q):
#elem min max mean median n50 sum 0cvg 8809 1 34854 119 38 438 1050135
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0312_edena/
SSAKE/Edena/Velvet summary
host: sycamore.umiacs.umd.edu (Linux 64)
program version run-time SSAKE 3.0 8459.472u 31.849s 5:08:59.82 Velvet 0.5.05 528.553u 42.123s 10:36.12 Edena 2.11 1711.702u 11.351s 28:58.44
Input: 8627900 33 bp Solexa reads(2 lanes)
Output: contigs+singletons
Contig statistics:
program #seqs min max mean med n50 sum SSAKE 185030 34 5490 77 44 87 14287079 Velvet 10684 45 16239 640 363 1184 6841458 Edena 11180 100 11300 552 356 837 6175460
200bp+ contig statistics:
program #seqs min max mean med n50 sum SSAKE 12532 200 5490 486 385 549 6090567 Edena 8316 200 11300 692 488 902 5759209 Velvet 7382 200 16239 877 586 1252 6474426
Singleton statistics:
program #seqs SSAKE 3164495 Velvet 1273164 Edena 3955865
Gene assemblies
Dan Sommer reduced number of contigs >=200bp from 306 to 120 /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Annotation/PAb1.fasta
Contig stats: desc #elem min max mean stdev sum contigs 120 212 512638 51438 81999 6172675
Average Gap: 105 nt bases Median Gap: 14 nt bases Largest Gap: 1095 nt bases
927 singletons assembled
Contig Assemblies
Comparative
AMOScmp on the velvet contigs (PA14 ref) *
- Use only velvet ctgs
- This assembly will have some of the repeats collapsed
All:
#elem min max mean median n50 sum ctg 3046 45 27957 1928 1101 3744 5873032 singl 989 45 16239 836 292 2221 826462 all 4035 45 27957 1660 850 3573 6699494 breaks 270
200 bp+:
#elem min max mean median n50 sum ctg 2632 200 27957 2213 1390 3798 5824679 singl 586 200 16239 1344 721 2352 787686 all 3218 200 27957 2055 1243 3631 6612365 breaks 265
Reduced the number of contigs from 10684 to 4035 !!!
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0311_velvet-24/AMOScmp-PA14
AMOScmp on the velvet contigs & velvet singletons (PA14 ref) *
- Use singletones from velvet ctgs as well
- This assembly will have some of the repeats collapsed
#elem min max mean median n50 sum all 2383 33 177630 2592 72 45071 6177155 200bp+ 513 200 177630 11778 738 45351 6042047 all-velvet 334 59 177630 17920 7295 45351 5985407 # ctgs that contain at least 1 velvet ctg singl-velvet 984 45 16239 837 292 2221 823862 # velvet ctgs left as singletons singl-read 393416 33 45 33 33 33 12983724 # original Solexa reads not assembled
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0311_velvet-24/AMOScmp-PA14.singletons
AMOScmp on the velvet contigs (PAO1 ref)
All:
#elem min max mean median n50 sum ctg 2982 45 27957 1928 1124 3652 5749909 singl 1255 45 16239 764 247 2275 958885 all 4237 45 27957 1583 773 3440 6708794 breaks(PAO1) 271
200 bp+:
#elem min max mean median n50 sum ctg 2610 200 27957 2186 1380 3675 5705769 singl 707 200 16239 1283 674 2398 906818 all 3317 200 27957 1994 1205 3494 6612587 breaks(PAO1) 266
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0311_velvet-24/AMOScmp-PAO1
AMOScmp on the velvet contigs (PA14 & PA2192 ref)
Input: above contigs & singletons
All: desc #elem min max mean stdev sum ctg 2841 45 34374 2022 2570 5744186 singl 751 45 17475 1251 2264 939443 # 593 are velvet original ctgs all 3592 45 34374 1861 2529 6683629
200bp+: desc #elem min max mean stdev sum ctg 2489 200 34374 2291 2638 5701588 singl 456 200 17475 2001 2648 912503 all 2945 200 34374 2246 2641 6614091
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0311_velvet-24/AMOScmp-PA14-PA2192
AMOScmp on the velvet contigs (PA14, PA2192 & PACS2 ref)
Input: above contigs & singletons
All: desc #elem min max mean stdev sum ctg 2759 45 28263 2029 2554 5597067 singl 725 45 34374 1495 2744 1084142 # 493 are velvet original ctgs all 3484 45 34374 1918 2603 6681209
200bp+: desc #elem min max mean stdev sum ctg 2419 200 28263 2297 2618 5556096 singl 463 200 34374 2290 3170 1060442 all 2882 200 34374 2296 2714 6616538
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0311_velvet-24/AMOScmp-PA14-PA2192-PACS2
AMOScmp on the velvet contigs (PA14, PA2192, PACS2 & PAO1 ref)
Input: above contigs & singletons
All: desc #elem min max mean stdev sum ctg 2696 45 24046 2028 2510 5468683 singl 753 45 34374 1609 3050 1211809 # 483 are velvet original ctgs all 3449 45 34374 1937 2643 6680492
200bp+: desc #elem min max mean stdev sum ctg 2369 200 24046 2292 2569 5429268 singl 487 200 34374 2439 3527 1187586 all 2856 200 34374 2317 2756 6616854
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0311_velvet-24/AMOScmp-PA14-PA2192-PACS2-PAO1
AMOScmp/minimus on the AMOScmp (PA14 ref) & velvet "unique" contigs
Input:
AMOScmp PA14 ref assembly (2) contigs velvet "unique: contigs !!! No singletons
AMOScmp2 steps:
- Align AMOScmp & velvet contigs (nucmer -c 40) to each other
- Identify velvet contigs CONTAINED in AMOScmp contigs(max trimming of 20)
- Remove CONTAINED contigs from the velvet set
- Create alignment file based to the AMOS.scaff file (if nucmer is repeat contigs are collapsed/no placed exactly)
- Align velvet contigs to reference using nucmer ; filter only the CONTAINED ones
- Merge AMOS & velvet alignments
- Run "casm-layout -S -r ..." ; if -S is not used, many AMOS contigs are not placed
- Run "make-consensus ..."
- All AMOS input contigs should be part of the new assembly
- If the velvet CONTAINED alignments are not filtered, some end conflicts might cause some of the AMOS contigs end up as singletons
Input:
All:
desc #elem min max mean stdev sum
AMOS 1843 20 127984 3350.38 10776 6174765
velvet-uniq 1189 45 16239 997.85 1485.34 1186452 # 462 aligned to PA14 ref (nucmer -c 40);
# 284 CONTAIEND in the ref
# 178 (462-284) partially aligned :
# 58 have 2+ alignments
# 120 have 1 alignment
# 727 (1189-462) not aligned to the ref
all 3032 20 127984 2427.84 8530.33 7361217
200bp+: desc #elem min max mean stdev sum AMOS 542 200 127984 11245 17520 6094904 velvet-uniq 848 200 16239 1354.36 1627.86 1148499 # 422 aligned to PA14 ref (nucmer -c 40); 249 CONTAIEND in the ref; 426 not aligned to the ref all 1390 200 127984 5211.08 12019.6 7243403
Output:
All:
desc #elem min max mean stdev sum
ctg 1327 20 221554 4644.9 19079. 6163795 # two of them(530,531) contain only velvet seqs
# 1210 contain only AMOS seqs ; 11 contain 2 AMOS seqs (negative gap got merged)
# 115 contain both AMOS & velvet seqs
singl 817 45 16274 1053.94 1719.12 861076 # all but one(807) are velvet : see .conflict file
# 170 out of 178 partially aligned (see above) is a singleton
all 2144 20 221554 3276.52 15146. 7024871
200bp+: desc #elem min max mean stdev sum ctg 282 203 221554 21651 36731 6105733 singl 571 200 16274 1461.55 1917.62 834546 all 853 200 221554 8136.31 23189 6940279
minimus steps:
- Run minimus2 pipeline on the above contigs(1327) and singletons(817) ; Only uses contig:singletion overlaps (not all:all)
$ ~dpuiu/bin/minimus2 -D BREAKID=1327 -D OVERLAP=40 Pa
min OVL=40 : use contig:singleton alignments
Output:
All: desc #elem min max mean stdev sum ctg 180 135 253489 22817.2 45339 4107096 # 218 of the PA14 unaligned velvet contigs assembled singl 1377 20 153036 1996.79 10534 2749587 all 1557 20 253489 4403.77 19467 6856683 200bp+: desc #elem min max mean stdev sum ctg 174 210 253489 23598.3 45919 4106116 singl 488 200 153036 5519.51 17154 2693524 all 662 200 253489 10271.3 28846 6799640
min OVL=20 : use contig:singleton alignments (not significantlty better than ,on OVL=40)
Output:
All: desc #elem min max mean stdev sum ctg 279 63 253489 15557 37925 4340305 # 321 of the PA14 unaligned velvet contigs assembled singl 972 20 153036 2576 12252 2504000 all 1251 20 253489 5471 21580 6844305
200bp+: desc #elem min max mean stdev sum ctg 263 214 253489 16495 38868 4338061 singl 393 200 153036 6261 18680 2460755 all 656 200 253489 10364 28954 6798816
min OVL=20 : use contig:singleton & singleton:singleton alignments
All: desc #elem min max mean stdev sum ctg 277 103 253489 15983 37991 4427292 # 556 of the PA14 unaligned velvet contigs assembled singl 808 20 153036 2986 13439 2412439 # 260 of them are velvet original contigs all 1085 20 253489 6304 23111 6839731
200bp+: desc #elem min max mean stdev sum ctg 263 222 253489 16826 38812 4425275 singl 298 201 153036 7971 21242 2375456 # 165 of them are velvet original contigs all 561 201 253489 12123 31042 6800731
min OVL=20 : use contig:singleton & singleton:singleton alignments & adjacent contig ovelaps (best)
All:
desc #elem min max mean stdev sum
ctg 317 47 253489 16450 40806 5214579
singl 703 20 112908 2324 11332 1633528 # 335 are original velvet contigs
# 29 are velvet contigs partially aligned
all 1020 20 253489 6714 25448 6848107
200bp+: desc #elem min max mean stdev sum ctg 291 200 253489 17909 42289 5211547 singl 247 201 112908 6480 18431 1600539 # 186 are original velvet contigs all 538 200 253489 12662 33969 6812086
Locations:
/fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/AMOScmp-velvet /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/AMOScmp-velvet/AMOScmp2 /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/AMOScmp-velvet/AMOScmp2.not_CONTAINED
AMOScmp/minimus on the AMOScmp (PA14 ref) & velvet contigs *
- Use all velvet contigs
- Very similar with the previous case (the velvet CONTAINED contigs were discarded at the beginning)
AMOScmp2
Input:
All: desc #elem min max mean stdev sum AMOS 1843 20 127984 3350 10777 6174765 velvet 10684 45 16239 640 825 6841458 all 12527 20 127984 1039 4311 13016223 200bp+: desc #elem min max mean stdev sum AMOS 542 200 127984 11245 17521 6094904 velvet 7382 200 16239 877 896 6474426 all 7924 200 127984 1586 5344 12569330
Output:
All: desc #elem min max mean stdev sum ctg 1361 20 221563 4536 19428 6173665 singl 838 45 22884 1064 1868 891463 all 2199 20 221563 3213 15418 7065128
200bp+: desc #elem min max mean stdev sum ctg 299** 203 221563 20448 37378 6113919 singl 576 200 22884 1499 2114 863431 all 875 200 221563 7974 23668 6977350
minimus2 on AMOScmp2 output
All: desc #elem min max mean stdev sum ctg 284 47 274913 18279 46193 5191268 singl 1001 20 133259 1666 9754 1667778 all 1285 20 274913 5338 24330 6859046
200bp+: desc #elem min max mean stdev sum ctg 246 210 274913 21088 49046 5187663 singl 285 201 133259 5696 17670 1623459 all 531 201 274913 12827 36583 6811122
Locations:
/fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/AMOScmp-velvet/AMOScmp2 /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/AMOScmp-velvet/AMOScmp2/minimus2/
AMOScmp/minimus on the AMOScmp PA14 & PAO1 contigs
AMOScmp
Input:
All:
desc #elem min max mean stdev sum
PA14 1843 20 127984 3350 10777 6174765
PAO1 1961 20 46414 3065 5251 6010800 # 1655 aligned
# 1312 CONTAINED in PA14 contigs
# 343 aligned but not CONTAINED in PA14 ctgs; 36 have 2+ alignments
# 306 not aligned
all 3804 20 127984 3203 8396 12185565
200bp+: desc #elem min max mean stdev sum PA14 542 200 127984 11245 17521 6094904 PAO1 1166 203 46414 5108 6007 5955960 all 1708 200 127984 7056 11405 12050864
Output:
All: desc #elem min max mean stdev sum ctg 1585 20 159598 3896 15496 6174745 singl 266 20 26709 1900 4486 505395 # all singletons are PAO1 all 1851 20 159598 3609 14456 6680140
200bp+: desc #elem min max mean stdev sum ctg 356 200 159598 17137 29063 6100717 singl 75 215 26709 6631 6361 497316 all 431 200 159598 15309 26837 6598033
minimus2 on the above contigs and singletons:
Output:
All: desc #elem min max mean stdev sum ctg 246 30 266798 18632 44344 4583528 singl 1138 19 126780 1569 9194 1785442 # 161 are original PAO1 contigs all 1384 19 266798 4602 21458 6368970
200bp+: desc #elem min max mean stdev sum ctg 139 202 266798 32900 54951 4573157 singl 152 201 126780 11405 22894 1733533 # 23 are original PAO1 contigs all 291 201 266798 21672 42726 6306690
AMOScmp2 on the AMOScmp PA14 contigs & PA14 genes *
INPUT:
#elem min max mean median n50 sum AMOScmp 1843 20 127984 3350 63 30995 6174765 AMOScmp(200bp+) 542 200 127984 11245 3263 31106 6094904 genes 5892 72 15639 992 861 1206 5846175
OUTPUT:
#elem min max mean median n50 sum all 650 20 187557 9954 846 60335 6470391 200bp+ 462 201 187557 13977 1578 60335 6457375 0 singletons !!!
Contigs that contain at least 1 AMOScmp contig & 1 gene: 356
#elem min max mean median n50 sum "singletons" ctg-all 356** 111 187557 17861 2692 63102 6358519 ctg-200bp+ 353 217 187557 18012 2723 63102 6358095 ctg-AMOS 356 1 85 5 2 9 1653 190 ctg-genes 356 1 160 16 2 58 5781 111 ** that many genes from PA14 seem to miss from PAb1
Contigs that contain at least 1 AMOScmp contig(>100bp) & 1 gene: 218
#elem min max mean median n50 sum ctg-all 218** 158 187557 28371 13008 66709 6184790 ctg-200bp+ 217 305 187557 28501 13008 66709 6184632 ctg-AMOS 218 1 19 3 2 4 685 ctg-genes 218 1 160 26 12 59 5632
Contigs that contain at least 1 AMOScmp contig(>200bp) & 1 gene: 206
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/AMOScmp-gene/AMOScmp2
AMOScmp2 on the AMOScmp PA14 contigs , PA14 genes * & velvet contigs
INPUT:
#elem min max mean median n50 sum AMOScmp 1843 20 127984 3350 63 30995 6174765 AMOScmp(200bp+) 542 200 127984 11245 3263 31106 6094904 genes 5892 72 15639 992 861 1206 5846175 velvet 10684 45 16239 640 363 1184 6841458
OUTPUT:
#elem min max mean median n50 sum
all 552 20 337673 11718 663 133802 6468490
200bp+ 377 201 337673 17126 1155 133802 6456628
singl 1006 45 16239 869 306 2308 874312 # all but 2 are velvet ctgs;
# 232 velvet singletons have annotated alignments to the AMOScmp ctgs
About half of the contigs end up in genes
Contigs that contain at least 1 gene & (1 AMOS ctg >=100bp or 1 velvet ctg): 135
#elem min max mean median n50 sum ctg-all 135** 163 337673 45819 14232 141346 6185574 ctg-200bp+ 134 305 337673 46160 16317 141346 6185411 ctg-AMOS100-vel 135 1 508 77 28 215 10356 ctg-genes 135 1 287 42 12 126 5631
Contigs that contain at least 1 gene & 1 velvet ctg: 104 Contigs that contain at least 1 gene , 1 velvet ctg & 1 AMOScmp ctg : 104 (same number)
#elem min max mean median n50 sum ctg-all 104** 163 337673 58949 32961 141346 6130650 ctg-200bp+ 103 747 337673 59519 32961 141346 6130487 ctg-AMOS 1085 ctg-velvet 9667 ctg-genes 5572
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/AMOScmp-velvet-gene/AMOScmp2
AMOScmp on velvet & edena contigs (PA14 ref)
Input:
#elem min max mean median n50 sum velvet 10684 45 16239 640 363 1184 6841458 edena 11180 100 11300 552 356 837 6175460 all 21864 45 16239 595 359 992 13016918
PA14 0cvg regions (nucmer -c 40; delta-filter -q):
#elem min max mean median n50 sum velvet 2388 1 31832 219 24 6434 522048 edena 8809 1 34854 119 38 438 1050135 all 2129 1 31832 234 22 7529 498077
Output:
All:
#elem min max mean median n50 sum
ctg 2782 45 27957 2141 1207 4246 5955924
singl 1874 45 16239 839 343 1972 1572938
all 4656 45 27957 1617 727 3749 7528862
breaks 492
200 bp+:
#elem min max mean median n50 sum
ctg 2424 200 27957 2439 1505 4288 5912995
singl 1233 200 16239 1217 677 2121 1500822
all 3657 200 27957 2027 1122 3816 7413817
breaks 478
AMOScmp on velvet contigs (PA14 gene ref) *
Contig stats:
#elem min max mean median n50 sum nucmer (-l 20,-c 65) 3137 65 3615 490 370 705 1535656 promer (-l 6, -c 10) 3622 61 6125 623 448 922 2255378
Only the contigs CONTAINED in the genes get assembled; BEGIN/END are discarded
Locations:
/fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0311_velvet-24/genes/AMOScmp-nucmer /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0311_velvet-24/genes/AMOScmp-promer
De-novo
minimus on velvet contigs (OVL=40)
All:
#elem min max mean median n50 sum ctg 179 45 1495 75 45 45 13336 singl 10324 45 16239 661 384 1188 6819550 all 10503 45 16239 651 374 1185 6832886 breaks 257
200 bp+:
#elem min max mean median n50 sum ctg 6 227 1495 836 1129 1495 5015 singl 7375 200 16239 877 586 1252 6469371 all 7381 200 16239 877 586 1252 6474386 breaks 251
minimus on velvet contigs (OVL=20)
All:
#elem min max mean median n50 sum ctg 1812 47 23446 1621 1164 2366 2936650 singl 6135 45 15342 626 357 1136 3839779 all 7947 45 23446 853 471 1574 6776429 breaks(PA14) 299
200 bp+:
#elem min max mean median n50 sum ctg 1746 201 23446 1677 1211 2374 2927255 singl 4246 200 15342 851 568 1216 3615358 all 5992 200 23446 1092 702 1651 6542613 breaks(PA14) 293
minimus1 on velvet contigs (OVL=20)
All:
#elem min max mean median n50 sum ctg 2046 73 23446 1628 1157 2396 3330276 singl 5658 45 15342 609 344 1144 3445004 all 7704 45 23446 879 489 1660 6775280 breaks(PA14) 568
200 bp+:
#elem min max mean median n50 sum ctg 1978 201 23446 1678 1205 2396 3320032 singl 3786 200 15342 857 575 1222 3243138 all 5764 200 23446 1139 737 1729 6563170 breaks(PA14) 559
If OVL is dropped to 16, fewer contigs get merged => fewer breakpoints
minimus2 on the AMOScmp PA14 & PAO1 contigs
- Use PA14-PA14 (5 bp, adj ctg ends) , PA14-PAO1 (20 bp, contig ends) , PAO1-PAO1 (5 bp, adj ctg ends) overlaps
All:
#elem min max mean median n50 sum ctg 441 43 223777 13972 294 86090 6161798 singl 1227 20 18974 237 42 6391 290505 all 1668 20 223777 3868 52 78110 6452303 breaks(PA14) 130
200 bp+:
#elem min max mean median n50 sum ctg 245 202 223777 25080 5100 86090 6144514 singl 107 210 18974 2180 547 8193 233281 all 352 202 223777 18119 1678 78110 6377795 breaks(PA14) 121
Location:
/fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/AMOScmp-PA14-PAO1/minimus2
minimus2 on AMOScmp PA14 contigs and PA14 genes (OVL=20)
INPUT:
#elem min max mean median n50 sum AMOScmp 1843 20 127984 3350 63 30995 6174765 genes 5892 72 15639 992 861 1206 5846175
OUTPUT:
#elem min max mean median n50 sum singl ctg-all 278** 158 170499 22136 10332 52297 6153935 ctg-200bp+ 277 315 170499 22216 10332 52297 6153777 ctg-AMOS 278 1 23 2 1 3 604 1239 ctg-genes 278 1 157 20 10 49 5582 310
minimus2 on velvet contigs and PA14 genes (OVL=20)
- min %id: = 94 (set from cmd line)
INPUT
#elem min max mean median n50 sum gene 5892 72 15639 992 861 1206 5846175 velvet 10684 45 16239 640 363 1184 6841458
OUTPUT (nucmer,OVL=20)
#elem min max mean median n50 sum ctg 923** 181 66497 6728 4737 10446 6210244 singl-gene 352 93 7347 979 777 1272 344478 singl-velvet 1144 45 16239 580 214 1603 663940 all 2419 45 66497 2984 901 9049 7218662
Best alignments:
show-coords Pa.filter-1.delta | ~/bin/AMOS/mummerAnnotate.pl -all | awk '{print $20}' | count.pl
[CONTAINED] 5306
[END] 3482
[BEGIN] 3453
[CONTAINS] 1977 # ??? why only 1977 velvet ctgs contain full genes; genes are not repetitive; even worse for edena
[PARTIAL] 168
[IDENTITY] 1
Total 14387
0cvg regions in the genes
#genes #elem min max mean median n50 sum 1391 1940 1 1762 36 18 67 69336
=>4166 out of 5557 assembled genes are fully covered by velvet ctgs
Location: /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0311_velvet-24/genes-PA14/minimus2-nucmer.OVL20
minimus2 on velvet contigs and Ps genes (OVL20)
- min %id: = 74 (nucmer default)
- avg %id: = 84
min%id overlaps 74 1162 # all considered 85 415 90 110
INPUT
#elem min max mean median n50 sum gene 5478 75 18825 996 852 1227 5456880 velvet 10684 45 16239 640 363 1184 6841458
OUTPUT
#elem min max mean median n50 sum "singl" ctg 652** 323 18825 2150 1768 2451 1401627 !!! 1.4M of genome gets covered ctg-gene 757 96 18825 1271 1122 1455 962199 4721 ctg-velvet 998 45 11120 938 558 1767 935636 9686
minimus2 on velvet contigs and Ps genes (OVL36: promer -l 6 -c 12)
min%id overlaps genes velvet 22 8496 2985 4827 #default 50 7209 2813 4495 60 5948 2571 4087 70 4361 2122 3269 74 3700 1880 2827 80 2615 1407 2036 85 1714 956 1311 90 976 516 659
min%id=74 =>
* 3700 overlaps * 2169 Ps genes * 3206 velvet contigs
make-consensus error
#elem min max mean median n50 sum "singl" ctg 929** 206 16291 2012 1669 2335 1869539 !!! 1.8M of genome gets covered ctg-gene 991 111 9522 1118 990 1272 1107648 4487 # 2169-991 genes had overlaps but were not assembled ctg-velvet 1032 45 16239 1199 910 1742 1237453 9652 # 3206-1032 velvet contigs had overlaps but were not assembled
!!! Slightly more genes/velvet contigs get assembled with promer than nucmer
Final assembly (v3)
Merged some of the 120 AMOScmp contigs from v2 using velvet contigs. Grouped overlapping contigs and used minimus to merge them.
Contig length stats: desc #ctgs min max mean stdev sum final3 512 200 512638 13099.41 53526.8 6706902 AMOScmp3 76 212 512638 82763.22 117199.28 6290005 novo3 436 200 10493 956.18 1336.44 416897 final3-10KB+ 49 10493 512638 127086.12 125757.98 6227220
Comment:
~ 53-74 (out of 436) the velvet denovo that contigs align to PA14.
Length statistics:
#ctgs min max mean stdev sum
CONTAINED 53 204 7228 514.81 978.25 27285
ALL 74 204 7228 622.85 926.13 46091
Locations:
/fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/final3/ /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Assembly/2008_0311_velvet-24/nucmer/merge/
Annotation
5769 genes /fs/szasmg2/Bacteria/Pseudomonas_aeruginosa/Annotation/merge/gene_list.ptt