Pseudodomonas syringae
Pseudomonas syringae pv. tomato str. DC3000
Originally sequenced and finished at TIGR: published Sept 2003
Data
NCBI
AA: no assembly TA 80,959 reads Genome Project Taxonomy TaxId=223283
Chromosome + 2 plasmids:
Name Length %GC NC_004578.1 6,397,126 58.40 NC_004633.1 73,661 55.15 NC_004632.1 67,473 56.17
UNC: Jeff Dangl
New sequence:
* Solexa 3 lanes;
* 454 shotgun 1/4 Plate (250bp read);
* 454 paired ends 1/4 Plate :
* contain a 44 bp linker in the middle
* the linker sequence is: GTTGGAACCGAAAGGGTTTGAATTCAAACCCTTTCGGTTCCAAC
* there are some (not many) 454 paired end sequences that contain multiple instances of the linker (tandem): Example EUEIEUN01ANUGL_length=128_xy=0154_1891
UNC sequence data: (not avail any more?)
http://biology622.dhcp.unc.edu/~labweb/DCData/
UNC (e-mail):
* Theoretical minimum number of contigs we can obtain is 268 (our reads fail to cover 269 nucleotides). * Our de novo assembly spans the genome in 853 contigs totaling 6,313,026 bp. * 98.7% of the genome is covered by a contig; * 84% of the genome is covered by contigs 10,000 bp or greater. * The average gap size between contigs is 98 bp; * average contig size 7401 bp. * The N50 = 37,444 bp. * Our largest BAMBUS "scaffold" is 2,565,761 bp,
Data stats
. #elem min median max sum mean stdev n50
DC3000.format.454Reads.fna 123,992 38 86 329 15623908 126.01 58.89 142 DC3000 Paired End Reads (forward+linkerr+reverse)
DC3000.TCA.454reads.format.fna 77,466 35 244 371 18627363 240.46 26.85 245 DC3000 454 Reads
DC3000.reads.filtered.fasta 6,340,136 32 32 32 202884352 32 0 32 DC3000 Solexa Reads
DC3000Plasmids.fa 2 67473 73661 73661 141134 70567 3094 73661 Pseudomonas syringae pv. tomato DC3000 Plasmids
Psudomonas_syringae.fa 1 6397126 6397126 6397126 6397126 6397126 0 6397126 Pseudomonas syringae pv. tomato DC3000 reference
Quality values are missing for all data sets!!!
I assigned default qual=3 to all the base (.frg & .afg files)
Files location:
/fs/szasmg2/Bacteria/Pseudodomonas_syringae/Data /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly
Assemblies
CBCB
1. AMOSCmp
454 single reads + Solexa reads /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Solexa-454/2007_1009_AMOSCmp-relaxed 142 contigs (37 negative gaps, 89 positive gaps) No read trimming was done. AMOScmp used the following parameters: nucmer -c 20 casm-layout -t 20 -o 5 "-t 20" allows for 20 bp long dirty sequence ends which seem to solve the "low quality" problem. => 22 large contigs 454 single reads + 30 bp Solexa reads => 167 contigs , 49 negative gaps, 100 positive gaps 454 single reads + 25 bp Solexa reads => 293 contigs, 144 negative gaps, 131 positive gaps
2. AMOSCmp
454 single reads + Solexa reads + 454 paired ends Only the 454 paired ends that contain 1 single complete adaptor sequence were used (allmost all) /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Solexa-454-454p/2007_1011_AMOSCmp-relaxed-filtered 149 contigs; very similar to the prev ome
3. AMOSCmp (MAJORITY=50)
454 single reads + Solexa reads /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Solexa-454/2007_1015_AMOSCmp-relaxed-MAJORITY50 131 contigs (18 negative gaps) No read trimming was done. AMOScmp used the following parameters: nucmer -c 20 casm-layout -t 20 -o 5 -m 50 No read trimming was done. "-t 20" allows for 20 bp long dirty sequence ends which seem to solve the "low quality" problem. "-m 20" merges some contigs together => 10 large contigs
contig# len gc% 4 2290968 59.00 7 1817904 58.18 3 1405326 58.08 5 648413 58.48 2 192413 57.86 6 87152 58.02 131 71251 56.47 1 32939 54.86 130 29120 59.36 9 20309 53.56 95 3589 59.46
4. AMOSCmp
Sanger reads /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1011_AMOSCmp-relaxed Many miss-oriented mates in the 4.8M-5M region of the chromosome 22 contigs Chromosome Chromosome problem
{kind=link}
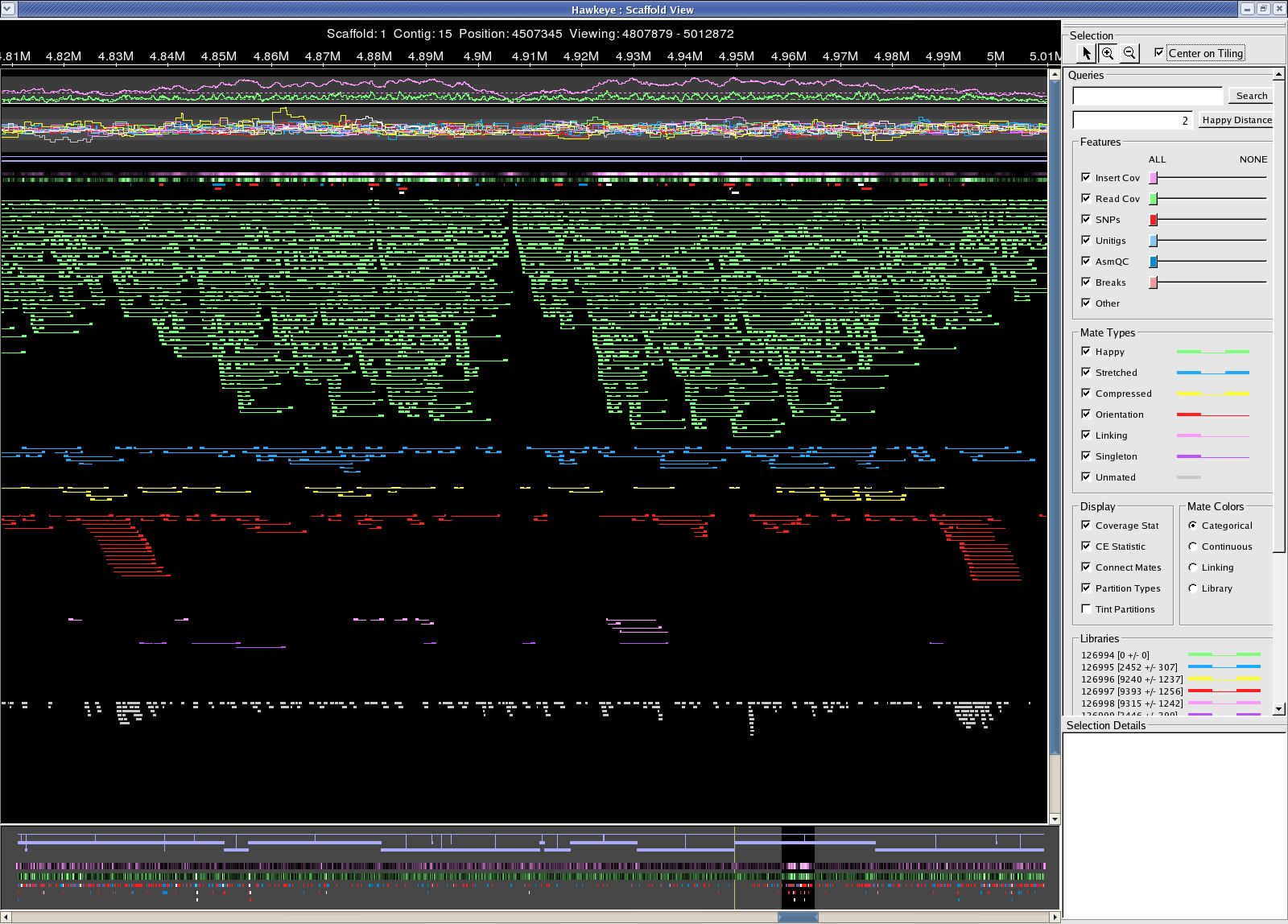{kind=link}
5. Celera 3.11
Sanger reads /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1011_WGA 22 scaff, 46 contigs, 181 degens Scaffold 7180000001443 looks circular: possible 163,074 bp plasmid aligns to 4.8M-5M "problem" region in the chromosome 7180000001443.png
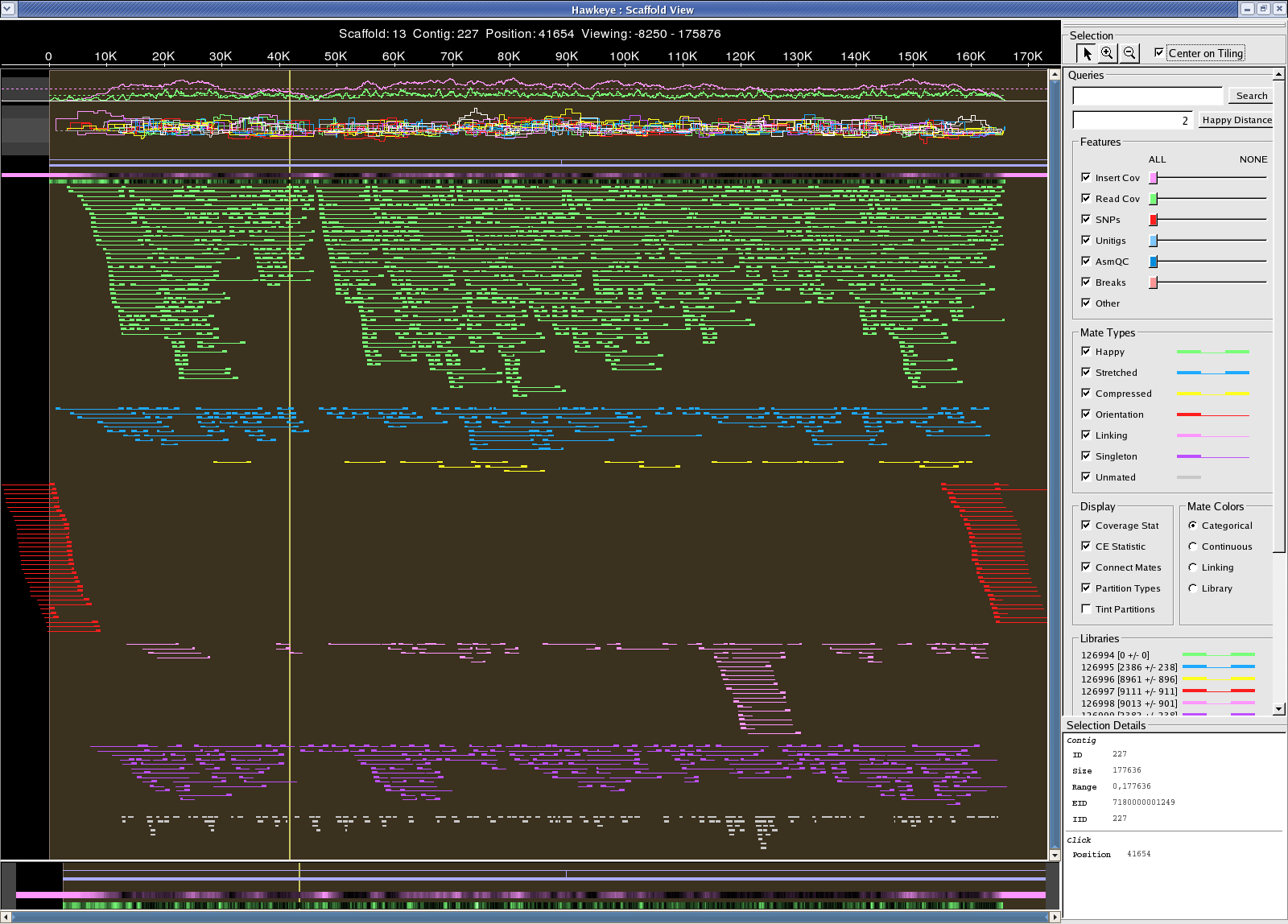{kind=link}
[S1] [E1] | [S2] [E2] | [LEN 1] [LEN 2] | [% IDY] | [LEN R] [LEN Q] | [COV R] [COV Q] | [TAGS]
===============================================================================================================================
1 175592 | 1 175592 | 175592 175592 | 100.00 | 175592 175592 | 100.00 100.00 | 7180000001443 7180000001443 [IDENTITY]
1 12519 | 163075 175592 | 12519 12518 | 99.98 | 175592 175592 | 7.13 7.13 | 7180000001443 7180000001443 [BEGIN]
163075 175592 | 1 12519 | 12518 12519 | 99.98 | 175592 175592 | 7.13 7.13 | 7180000001443 7180000001443 [END]
[S1] [E1] | [S2] [E2] | [LEN 1] [LEN 2] | [% IDY] | [LEN R] [LEN Q] | [COV R] [COV Q] | [TAGS] =============================================================================================================================== 4790727 4911492 | 120764 1 | 120766 120764 | 99.98 | 6397126 175592 | 1.89 68.78 | gi|28867243|ref|NC_004578.1| 7180000001443 4898971 4955870 | 175592 118697 | 56900 56896 | 99.98 | 6397126 175592 | 0.89 32.40 | gi|28867243|ref|NC_004578.1| 7180000001443
6. AMOSCmp (Chromosome+3 plasmids ref)
Sanger reads Reference=complete genome(chromosome+3 plasmids) use "circular contig" in Celera 3.11 assembly /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1012_AMOSCmp-relaxed-3plasmids 38 contigs: 15 for main chromosome, 1 for longer plasmid, 21 for shorter plasmid, 1 for "circular contig" The missoriented read pile corresponding to the chromosome (4. AMOSCmp of Sanger reads) has dissapeared AA ready for submission: /fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Sanger/2007_1012_AMOSCmp-relaxed-3plasmids/AA/umd-20071030-141700.tar.gz
Solexa assemblied for different read coverages
Location
/fs/szasmg2/Bacteria/Pseudodomonas_syringae/Assembly/Solexa/sample/ Several AMOScmp assemblies, using 100%, 90% ... 10% of the P. syringae Solexa reads. These would correspond to 30X, 27X, 24X .. 3X coverage The read sampling was done randomly. One sample set for each coverage.
Chromosome + 2 plasmids
qc stats for Solexa assemblies done at different coverage levels
cvg: 30,27,24...3 $ more contig.chromo.summary positiveGaps.chromo.summary :::::::::::::: contig.summary :::::::::::::: %reads #elem #elem0 #elem<0 min median max sum mean stdev n50 100 5502 0 0 32 338 32148 7296600 1326.17 2157.6 3714 90 6463 0 0 32 330 25252 7252304 1122.13 1799.43 3009 80 7570 0 0 32 303 20690 7209479 952.38 1487.03 2573 70 9030 0 0 32 309 26306 7170384 794.06 1219.53 1986 60 10571 0 0 32 295 22249 7124996 674.01 961.22 1608 50 12598 0 0 32 274 22204 7075934 561.67 767.55 1266 40 15343 0 0 32 252 9176 7011485 456.98 575.64 934 30 21248 0 0 32 202 7751 6931907 326.24 376.06 597 20 38702 0 0 32 117 3276 6807914 175.91 178.92 278 10 84545 0 0 32 56 2652 6267925 74.14 57.62 90 :::::::::::::: positiveGaps.summary :::::::::::::: %reads #elem #elem0 #elem<0 min median max sum mean stdev n50 100 117 10 0 0 22 3065 19625 167.74 418.75 1308 90 130 16 0 0 19 2100 19725 151.73 369.07 1211 80 142 18 0 0 15 2174 20034 141.08 361.86 1209 70 178 15 0 0 9 3417 20443 114.85 395.13 1823 60 263 35 0 0 6 3875 21161 80.46 345.97 1457 50 450 64 0 0 4 3398 22305 49.57 278.39 1823 40 1047 156 0 0 4 3398 26488 25.3 173.77 929 30 2915 446 0 0 4 3426 39094 13.41 115.74 104 20 11154 1324 0 0 5 3420 110485 9.91 57.22 19 10 44751 3321 0 0 9 3875 631930 14.12 35.45 25
Chromosome (only)
$ more contig.chromo.summary positiveGaps.chromo.summary :::::::::::::: contig.chromo.summary :::::::::::::: %reads #elem #elem0 #elem<0 min median max sum mean stdev n50 100 5352 0 0 32 387 18942 7152892 1336.49 2069.25 3674 90 6313 0 0 32 362 16470 7110882 1126.39 1721.34 2969 80 7411 0 0 32 322 15227 7069778 953.96 1436.49 2521 70 8865 0 0 32 324 14901 7032202 793.25 1154.9 1968 60 10406 0 0 32 304 10231 6988498 671.58 919.5 1586 50 12389 0 0 32 279 7246 6941706 560.31 733.75 1247 40 15131 0 0 32 256 5409 6879810 454.68 554.17 920 30 20998 0 0 32 204 4102 6801160 323.9 358.93 588 20 38368 0 0 32 117 2220 6680303 174.11 170.14 274 10 83839 0 0 32 56 762 6144687 73.29 51.16 89 :::::::::::::: positiveGaps.chromo.summary :::::::::::::: . #elem #elem0 #elem<0 min median max sum mean stdev n50 100 15 5 0 0 1 33 107 7.13 10.84 33 90 24 7 0 0 2 42 146 6.08 10.38 42 80 38 11 0 0 2 36 212 5.58 8.84 26 70 76 11 0 0 3 33 413 5.43 6.66 11 60 163 29 0 0 4 33 1016 6.23 7.04 13 50 347 60 0 0 3 49 1843 5.31 6.45 11 40 947 151 0 0 4 53 5709 6.03 7.18 12 30 2819 442 0 0 4 63 17882 6.34 7.34 12 20 11029 1320 0 0 5 610 88516 8.03 10.84 15 10 44485 3313 0 0 9 197 606841 13.64 15.05 24