Culex pipiens symbiont
Data Sources
NCBI:
- Genome Project
- TA : 7,379,314 traces (Sept 2007)
Sanger: Wolbachia pipientis endosymbiont of Culex quinquefasciatus
Old reference: file name: /fs/szasmg2/Culex_pipiens_symbiont/Sanger/Wb_Cq_061226.dbs
Top 10 seqs Name Length %GC culex173d08.p1k 1457497 34.17 culexbac1d10Bg07.p1k 24726 35.11 culex3d09.p1k 15587 21.81 culex166f03.q1k 13962 36.17 culex_1177_1189-1a02.w2k1177 13564 37.10 culex26b07.p1k 9245 35.53 culex174d04.p1k 8832 33.64 J28015Ag08.q1ka 7809 36.04 culex180e07.p1k 6960 36.59 culex53a02.p1k 5343 33.58 ...
New reference (12 sequences): file name: /fs/szasmg2/Culex_pipiens_symbiont/Sanger/Wb_Cq.dbs
All seqs:
Name Length %GC
1 culexbac1b5Ab03.q1k 1136301 34.17
2 culex161b01.q1k 346054 34.25
3 culex166f03.q1k 13962 36.17 share almost all sequence with culex161b01.q1k & 1996bp with culexbac1b5Ab03.q1k
subtotal(3) 1496317
4 culex49c07.p1k 9245 35.53 looks circular(misoriented mates at the ends); region 4979-6364 aligns to culexbac1b5Ab03.q1k 3 times
5 culex53a02.p1k 5343 33.58 ~ 1Kbp alignments to culexbac1b5Ab03.q1k & culex161b01.q1k
6 culex117e02.p2kA55 3501 33.10 contained (in 2 pieces) in culexbac1b5Ab03.q1k
7 culex141a08.q1k 1920 33.44 few hundred bp alignments to other culex* seqs
subtotal(7) 1516326
8 culex180e07.p1k 6960 36.62 "CONTAINED" culexbac1b5Ab03.q1k (surrogate in WGA)
9 culex5c05.p1k 15587 21.81 low GC%; no alignments to NC_002978 & NC_006833; best hit is Anopheles gambiae complete mitochondrial genome : 15363 bp (96% coverage, 86% max id)
10 culex14h11.p1k 3350 51.73 repeat (higher GC%): good cvg of culex 18SrRNA gene ; no alignments to NC_002978 & NC_006833
11 culex22h10.q1k 2148 54.89 repeat (higher GC%): some alignment to culex 118S rRNA ; no alignments to NC_002978 & NC_006833
12 culex166d08.p1k 2071 55.53 repeat (higher GC%): culex 18S rRNA & 28S rRNA ; no alignments to NC_002978 & NC_006833
total(12) 1546442
JCVI:
Articles
Other Strains (complete)
RefSeq GenBank Pub Length (Mbp) GC Prot RNAs Wolbachia endosymbiont of Drosophila melanogaster(TIGR) NC_002978 AE017196 1 1.26778 35.2% 1195 39 Wolbachia endosymbiont strain TRS of Brugia malayi srain wMel(NEB) NC_006833 AE017321 1 1.08008 34.2% 805 37 Wolbachia pipientis wPip(Sanger) NC_010981 AM999887 1 1.48246 34.2% 1275 37
!!! Wolbachia pipientis wPip(Sanger) = culex161b01.q1k(346,054) + N(102) + culexbac1b5Ab03.q1k(1,136,301)
Read Counts
query_tracedb "query count SPECIES_CODE='CULEX PIPIENS QUINQUEFASCIATUS'" # 7552113 : all traces query_tracedb "query count SPECIES_CODE='CULEX PIPIENS QUINQUEFASCIATUS' AND load_date >='09/01/2007'" # 172799 : new traces (all cDNA)
Assembly
Locations:
/fs/szasmg2/Culex_pipiens_symbiont/
2006_1226_WGA : initial assembly
Steps:
1. All cpqg reads have been downloaded from the TA (July 2006). The reads have been grouped by libraries and the clear range has been computed. There were 6.6M reads in the download compared with 7.3M now. Unfortunately I've only noticed this difference at the end of my experiment.
2. The Wolbachia endosymbiont of Culex quinquefasciatus assembly has been downloaded from the Sanger ftp site ( ftp://ftp.sanger.ac.uk/pub/pathogens/Wolbachia/Wb_Cq.dbs ) ; there are 95 sequences in this file. Most of them are very short. Below are listed the name,length & gc% of the longest 10: name length(bp) gc% culex173d08.p1k 1457497 34.17 culexbac1d10Bg07.p1k 24726 35.11 culex3d09.p1k 15587 21.81 culex166f03.q1k 13962 36.17 culex_1177_1189-1a02.w2k1177 13564 37.10 culex26b07.p1k 9245 35.53 culex174d04.p1k 8832 33.64 J28015Ag08.q1ka 7809 36.04 culex180e07.p1k 6960 36.59 culex53a02.p1k 5343 33.58
3. The cpqg random reads (clr only) have been aligned to symbiont sequences using nucmer (default parameters)
4. The nucmer output has been analyzed. It's been noticed that many of the short symbiont sequences (2-3KB in length) have a higher than expected number of alignments. To avoid the repeats I've selected only the reads that aligned to the longest 10 symbiont sequences (see above).
5. A 95% identity and minimum of 400 bp alignment thold has been used to
determine the symbiont reads. There were 29,110 unique reads (30,690
reads+mates) selected. Below is a per library breakdown (reads+mates):
MSC-CULEX-PIPIENS-QUINQUEFASCIATUS_01-G-CULEX-10KB 9581
MSC-CULEX-PIPIENS-QUINQUEFASCIATUS_06-G-CULEX-10KB 4549
G818P4 3784
G818P2 3478
G818P1 2238
G818F1 1283
MSC-CULEX-PIPIENS-QUINQUEFASCIATUS_02-G-CULEX-4KB 1156
MSC-CULEX-PIPIENS-QUINQUEFASCIATUS_03-F-CULEX-40KB 738
G818P3 723
MSC-CULEX-PIPIENS-QUINQUEFASCIATUS_07-G-CULEX-10KB 556
MSC-CULEX-PIPIENS-QUINQUEFASCIATUS_05-F-CULEX-40KB 327
MSC-CULEX-PIPIENS-QUINQUEFASCIATUS_04-F-CULEX-40KB 185
1099522705601 99
G809K1 89 : cDNA , should be removed
1099499586718 77
G772K1 12 : cDNA , should be removed
G771K1 10 : cDNA , should be removed
G766BES1 4 : BE library
1099641499000 2
6. The reads have been assembled using the runCA-OBT.pl script (default parameters). Most of the reads got assembled into 3 large scaffolds. There is mate pair evidence (outie mates) that the largest scaffold is circular.
All the scaffolds ens up in surrogates (20-50KB total surrogate length) Are there not enough BE to span the unique regions?
Cpqg.qc
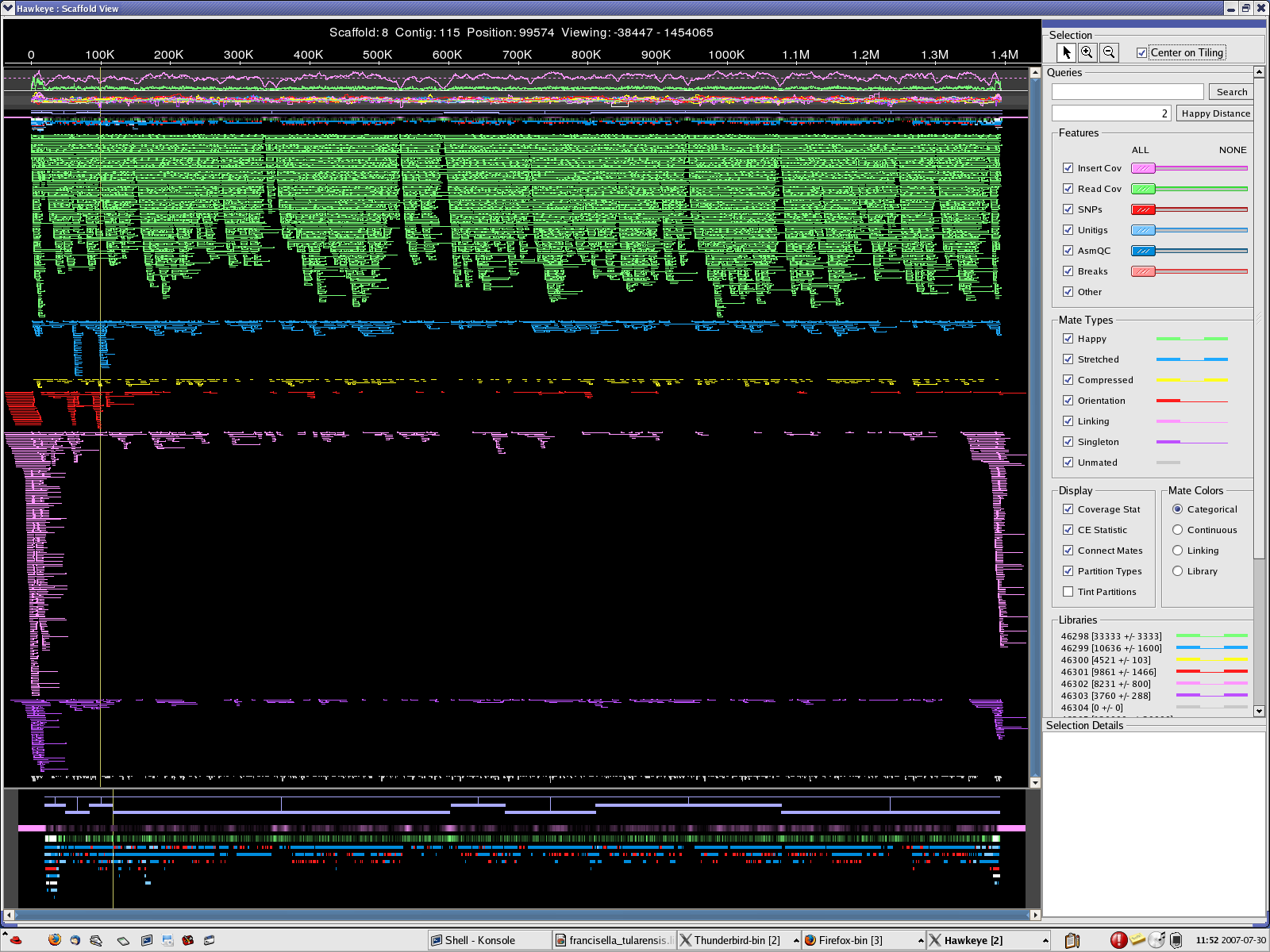
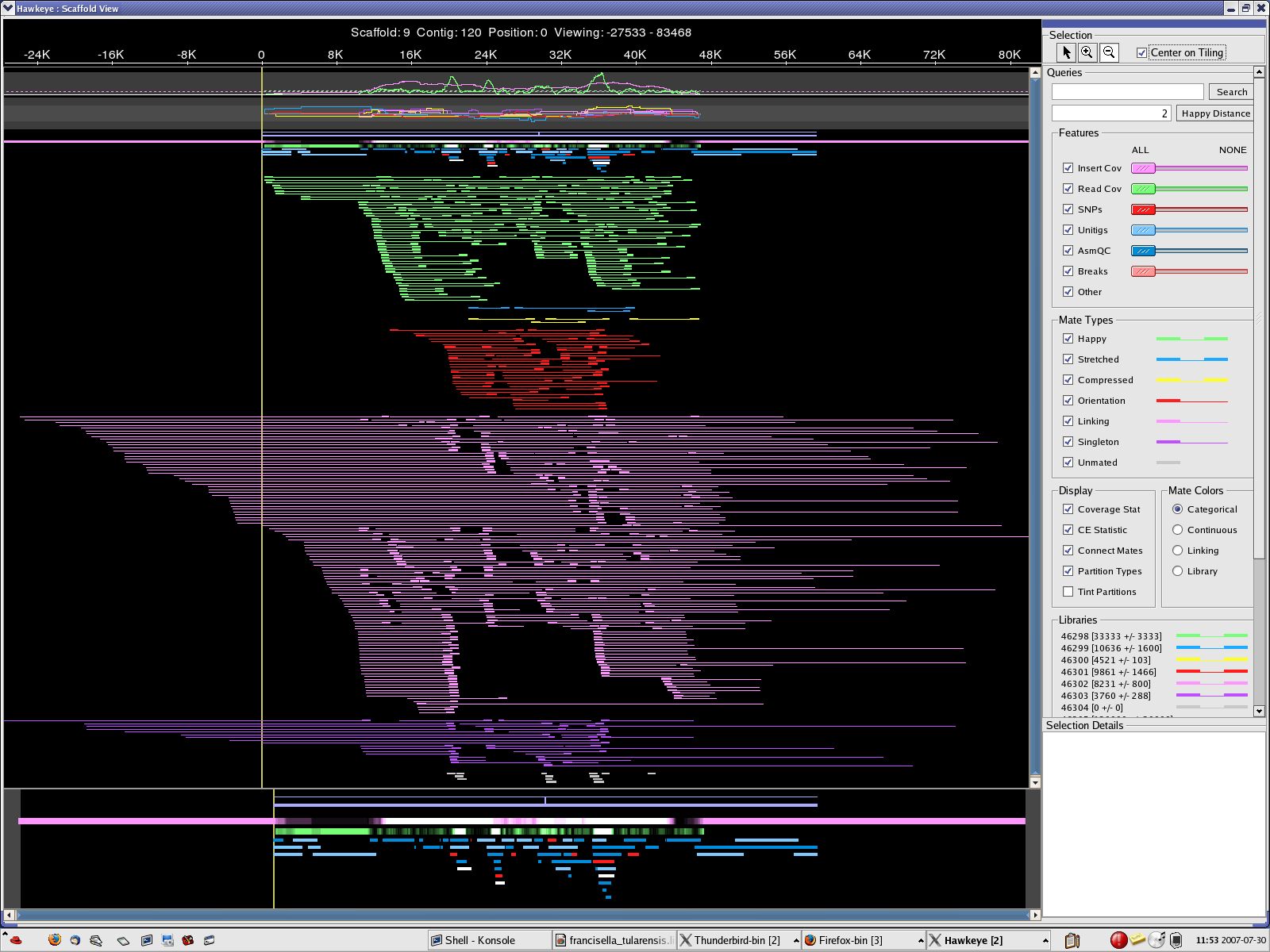
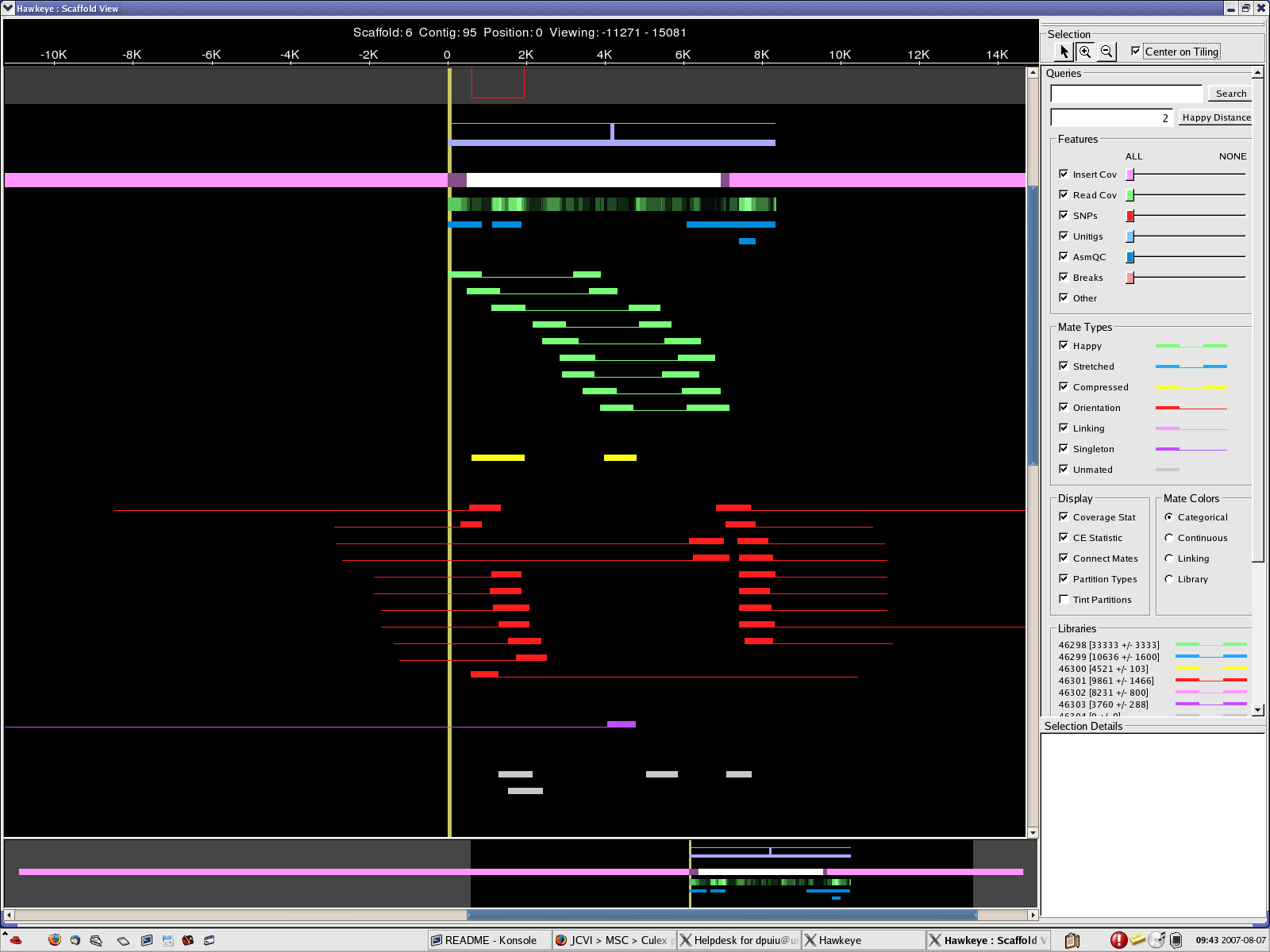
scaff_8 Longest scaff scaff_9 2nd longest scaff scaff_7 3rd longest scaff scaff_6 Small scaff that Looks circular
{kind=link}
{kind=link}
{kind=link}
{kind=link}
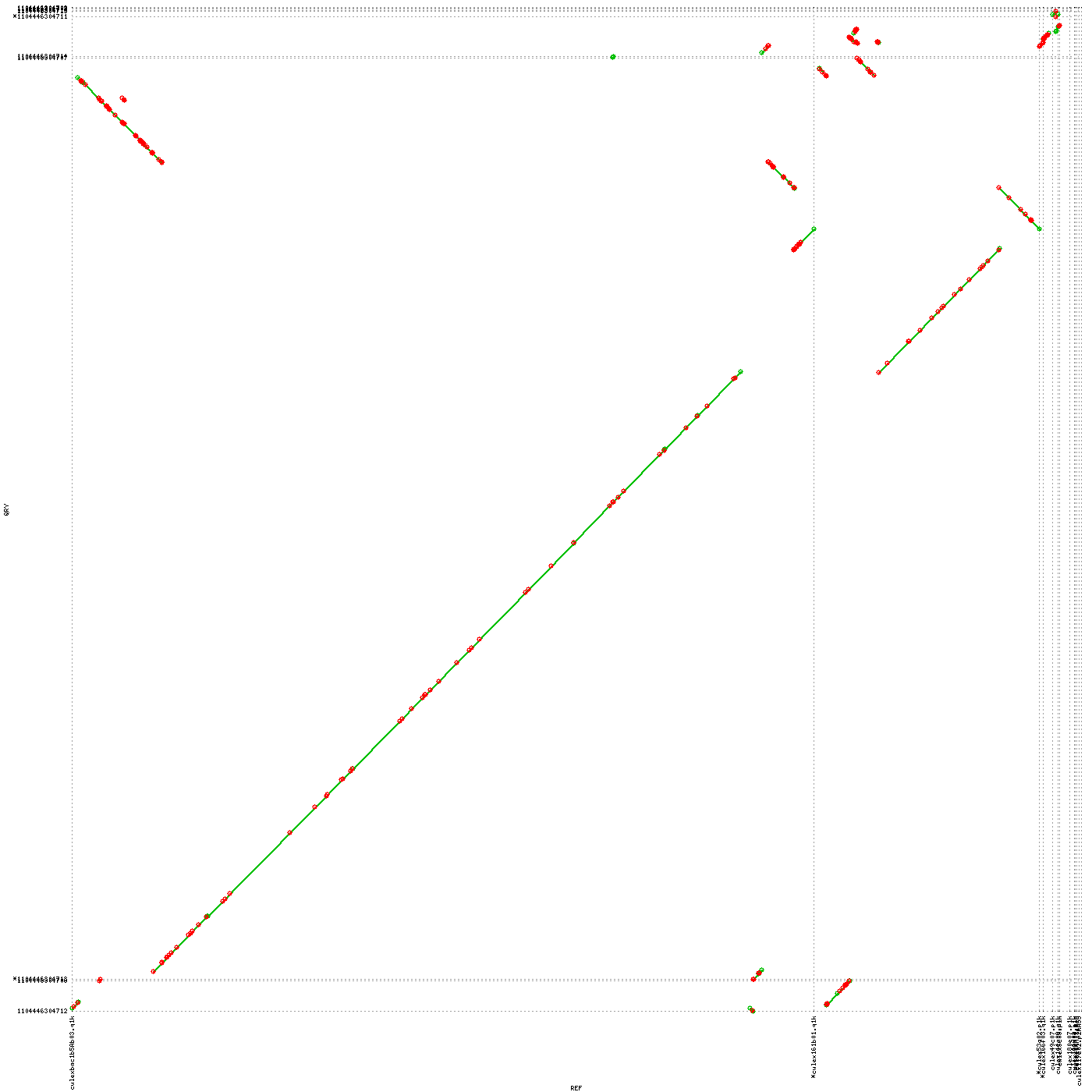
7. The scaffolds/contigs have been aligned to longest 10 Wolbachia endosymbiont sequences. Most of the long alignments were at over 99% identity. However, several large rearrangements have been noticed.
Wb_Cq-vs-scaff Reference vs scaff
{kind=link}
2007_0802_WGA-default : new assembly
Steps:
1. All Culex reads have been downloaded from TA . ~1M new reads sincd 2006_1226
2. The reads have been aligned to the new reference (exclude mito,repeats) using nucmer (default parameters)
3. A 95% identity and minimum of 400 bp alignment thold has been used to determine the symbiont reads. 3850 new reads & mates in addition to the previous ones were identified
4. 33,783 reads have been assembled using the runCA-OBT.pl script (default parameters). Cpqg.qc
Compared to the initial assembly, many metrics went down (TotalBasesInScaffolds,MaxBasesInScaffolds,MaxContigLength ...) TotalSurrogates & SurrogateInstances more than doubled
2007_0802_WGA-0.5E : error rate =0.5 % => more fragmented assembly
2007_0802_WGA-0.5M : genome size=1.5M => more TotalBasesInScaffolds but more unhappy mates
What to do next?
- use CA 5.1 (latest version)
- remove 958 cDNA's aligned to culex*
- increase utg error rate to from 1.5% to 2% (3% gave worse results than 2%)
- recruite reads that align to contig ends: some ends are repetitive => too many; others no alignments