Bumblebee: Difference between revisions
Jump to navigation
Jump to search
| Line 88: | Line 88: | ||
s_1 34944099 269156 9804247 24870696 6048164 | s_1 34944099 269156 9804247 24870696 6048164 | ||
s_2 32540640 91528 16181288 16267824 1061858 | s_2 32540640 91528 16181288 16267824 1061858 | ||
* Filtered read stats | |||
Revision as of 04:37, 18 March 2010
Data
- ~ 500B genome
- Complete mitochondrion genomes:
NC_011923.1 15468 14.67 Bombus hypocrita sapporoensis mitochondrion, complete genome NC_010967.1 16434 13.22 Bombus ignitus mitochondrion, complete genome only 88% identity; no rearrangements, only snps, short indels
Traces
- 7 pairs of data files (paired ends) : lanes 1..3,5..8 (lane 4 wasn't used)
Lane Insert ReadLen #Mates Coverage Comments 1 3K(2..6,avg 4K) 124 34,944,099 14X 865,687(1.2%) reads have qual==0 2 8K(7..9,avg 8K) 124 32,540,640 13X 3 400 124 34,745,750 # gDNA ; originally thought to be 500bp insert instead of 400 5 400 34,601,239 6 400 34,553,857 7 400 34,682,612 8 400 12,975,839
- Adaptors
>circularizarion CGTAATAACTTCGTATAGCATACATTATACGAAGTTATACGA >circularizarion.revcomp TCGTATAACTTCGTATAATGTATGCTATACGAAGTTATTACG >5 GATCGGAAGAGCGGTTCAGCAGGAATGCCGAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCG >3 CGGCATTCCTGCTGAACCGAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT
Tasks to figure out
- Erroneous reads/bases, which we need to correct or discard
- GC bias, so we can compute a-stats properly
- Redundancy in the long paired ends, which are lane 1 and lane 2.
- Used the 454 protocol to circularize the DNA for sequencing with the Illumina instrument.
- Some reads will begin in the circularization adaptor and thus will have only one usable read
- Some reads have a few bases of DNA sequence and hit the circularization adaptor right away
- Most reads will have at least 36bp from each end before hitting the adaptor.
- Many reads will not have any adaptor to trim (>125bp of DNA sequence at both ends of the adaptor)
- A small but significant number of reads from the 3kb and 8kb libraries are not recircularized.
- Thus their mate distance is +400bp rather than -3kb or -8kb.
- It's apparently the result of a faulty batch of cre recombinase. This causes problems with contiging and scaffolding.
- It is possible to remove these reads by removing
- mate pairs where neither read is trimmed (thus no adapter ligation may have occurred)
- mate pairs where one read begins with the adapter sequence.
- We are working on this, up to now our paired assembly stages have been disappointing.
- Matt's idea is to exclude all mate pair reads that don't have evidence of the linker with flanking useful sequence, as a way to avoid uncircularized molecules that will give misleading "mate pairs" only 400 bp apart.
- There has been no trimming of the adaptor, which is the 42 base 454 adaptor, so its presence can be used to indicate potentially good mate pairs.
- Even tossing half the mate pairs might not be a problem, as we have perhaps too many anyway.
- But you will also need to toss redundant mate pairs, and that will indeed reduce the total a lot.
- Just to be clear - the 500 base mate pairs should have no such problems, except that as Matt has found from his preliminary assembly, the mean fragment length is actually 400 bp rather than 500 bp (and the 3 and 8 kb PE reads are typically shorter than nominally given, e.g. more like 2.5 and 6 kb).
- You'll also need to throw out all the reads where one of the mates /starts/ with linker, I assume you'd do that anyway. We're also working on ways round this; one might be when we get a better assembly we'll find some better characteristic to filter the unrecircularized reads on.
Trimming
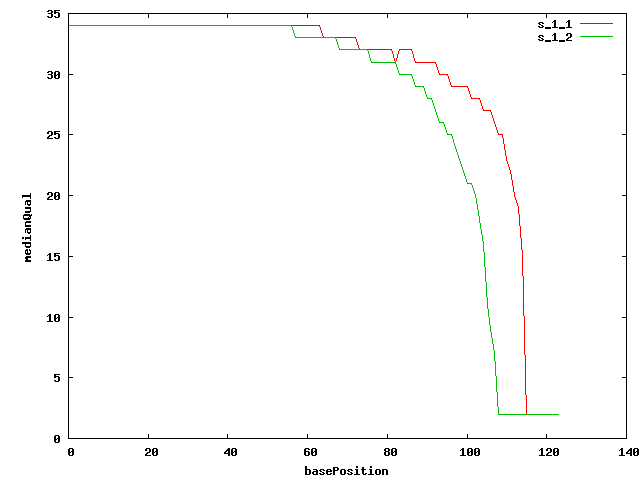{kind=link}
- Quality trimming: trim all bases with fastq quality eq "B" (0)
cat *fastq | ~/bin/fastq2clq.pl
- Adaptor trimming: Align all subsets to adaptors
C CGTAATAACTTCGTATAGCATACATTATACGAAGTTATACGA 3 CGGCATTCCTGCTGAACCGAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT 5 GATCGGAAGAGCGGTTCAGCAGGAATGCCGAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCG
id len gc% C 42 30.95 3 52 55.77 5 67 59.70
nucmer -l 8 -c 16 -b 32 -g 32 adaptors.seq ... delta-filter -l 16 -q ... cat *.filter-q.delta | ~/bin/delta2clr53.pl -5 5,3 ...
- Median adaptor/primer positions(5-3)
Lib adaptorPos 5'pos 3'pos s_1 34-75 0-36 0-19 s_2 2-40 0-36 0-19
- Mate stats
Lib #mates adaptor.bothMates adaptor.noMate adaptor.oneMate adaptor.oneMate(filtered) s_1 34944099 269156 9804247 24870696 6048164 s_2 32540640 91528 16181288 16267824 1061858
- Filtered read stats
- Other frequent kmers
26mer : ACGTTATAACGTATTACGTTATATGG -> revcomp -> CCATATAACGTAATACGTTATAACGT : ~10% of the traces 10mer : AAAAAAAAAA TTTTTTTTTT : ~32% of the traces 53mer: CGATTTCCATGGCGTCGTTTGAGGATTCCAATACGGCGAACCTGTTGTGAGTG : ~2% of the mito seqs (either begin or end); not present in the 2 complete mito's (probably ok)
- Location:
/fs/szattic-asmg4/Bees/Bombus_impatiens
D Kelly's trimming
438088072 total reads 109166398 reads were thrown away 148886138 reads were corrected and/or trimmed (to a min length of 30 bp)
Assembly
CA.bog
- Input
s_1 reads trimmed to 100bp & formatted by fastqToCA
- Spec file
unitigger = bog ovlOverlapper = mer obtMerThreshold = 400 ovlMerThreshold = 120 doOverlapTrimming = 0
- Unitigger : max utg len=852bp
- Consensus after unitigger : 3 out of 129 jobs failed
- Location
/scratch1/Bombus_impatiens/Assembly/31M
CA.utg
- Input
s_1 reads formatted by convert-fasta-to-v2.pl
- Spec file
unitigger = utg ovlOverlapper = ovl obtMerThreshold = 400 ovlMerThreshold = 120 doOverlapTrimming = 0 => reads < 64 bp atre trimmed by the gatekeeper; no need to trim