Dpuiu HTS
Summary
Vendor: Roche Illumina ABI
Technology: 454 Solexa GA SOLiD
-------------------- -------------------------------- ------------------
Platform: GS20 FLX Ti I II IIx HiSeq200 1 2 3
Reads: (M) 0.5 0.5 1.25 28 100 150 40 115 320
Fragment
Read length: 100 200 400 35 50 100 100 25 35 50
Insert: 0 (unmates)
Run time: (d) 0.25 0.3 0.4 3 3 5 8 6 5 8
Yield: (Gb) 0.05 0.1 0.5 1 5 15 200 1 4 16
Rate: (Gb/d) 0.2 0.33 1.25 0.33 1.67 3 25 0.34 1.6 2
Images: (TB) 0.01 0.01 0.03 0.5 1.1 2.8 1.8 2.5 1.9
PA Disk: (GB) 3 3 15 175 300 300 300 750 1200
PA CPU: (hr) 10 140 220 100 70 NA NA NA NA
SRA: (GB) 0.5 1 4 30 50 2.5 100 140 600
Paired-end
Read length: 200 400 2×35 2×50 2×100 2×25 2×35 2×50
Insert: (kb) 3.5 3.5 0.2 0.2 0.2 3 3 3
Run time: (d) 0.3 0.4 6 10 10 12 10 16
Yield: (Gb) 0.1 0.5 2 9 30 2 8 32
Rate: (Gb/d) 0.33 1.25 0.33 1.67 3 0.34 1.6 2
Images: (TB) 0.01 0.03 1 2.2 5.6 3.6 5 3.8
PA Disk: (GB) 3 15 350 500 500 600 1500 2400
PA CPU: (hr) 140 220 160 120 NA NA NA NA
SRA: (GB) 1 4 60 100 3.5 200 280 1200
Mate-pair
Vendor: Roche Illumina ABI
Technology: 454 Solexa GA SOLiD
-------------------- -------------------- ------------------
Year 2005 2007 2007
Amplification emulsion PCR bridge PCR emulsion PCR
Sequencing pyrosequencing sequencing-by-synthesis ligase-based sequencing
ErrorsRate/bp 5% 1% <0.1%
DominantError indels(esp homopolym) substitutions substitutions
Links
- Illumina technote
- Illumina technote
- Illumina technote data
- SAM format
- SAM tools
- DNAA
- [1] genome complexity graphs
- CA BOG paper
Modified Celera Assembler: the Celera Assembler software has modules for successive phases of assembly: pairwise overlap detection; initial ungapped multiple sequence alignments called unitigs; unitig consensus calculation; combination of unitigs with mate constraints to form contigs and scaffolds, which are ungapped and gapped multiple sequence alignments, respectively; and finally, scaffold consensus determination (Myers et al., 2000). Our approach to hybrid data assembly reuses the Celera Assembler scaffold and consensus modules. Independent of the hybrid problem, the scaffold module was revised to recover trimmed base calls confirmed by co-locating reads, and the consensus module was revised to determine alternate consensus sequences in regions of apparent polymorphism (Denisov et al., 2008). Our analysis narrowed the source of hybrid assembly problems to the overlap and unitig stages.
For speed, Celera Assembler relies on short exact matches between reads as seeds for overlap detection. Its exact-match algorithms were sensitive to the different proclivities for stutter observed between platforms. Stutter, that is, incorrect determination of the number of bases in homopolymer (single-letter) runs, is more prevalent in pyro reads than Sanger reads. We therefore modified the software to search for matches in compressed sequence, in which all single-letter repeats are reduced to a single base. The uncompressed sequence is consulted later before the seeds become overlaps.
Celera Assembler was sensitive to the different average read lengths between platforms. The shorter reads are more likely to be entirely contained within genomic repeats. Over-collapsed alignments of short repeat reads induce true and false overlaps to the interior of longer reads. Where the longer reads extend beyond the genomic repeats, they do not all overlap each other. The result is short reads with containment overlaps to multiple long reads that do not overlap each other. These overlap tangles were triggering Celera heuristics designed to detect mis-assembly, leading to unnecessarily short contigs.
Celera Assembler was also sensitive to the higher coverage typical of lower cost pyrosequencing. Higher coverage leads to increased collisions of reads with exactly the same prefix sequence. The assembler's arbitrary tie-breaking heuristics, sufficient for infrequent ties, had the potential to lead the assembler away from the global optimum in hybrid data. To address these problems we developed an aggressive approach to unitig construction that builds unitigs in greedy fashion, always following a read's best overlap (by an appropriate criterion), and ignoring contained reads at first. The aggressive unitigs initially incorporate mistakes that, ideally, are caught and corrected later by pattern analysis applied to best overlaps and mate constraints. High coverage could also increase the number of spurs, that is, reads with invalid sequence at one end. These seemed to contribute to fractured unitigs on hybrid data. We realized the software could turn higher coverage to its advantage by carefully trimming reads of unconfirmed sequence.'
- Substantial biases in ultra-short read data sets from high-throughput DNA sequencing
- Evaluation of next generation sequencing platforms for population targeted sequencing studies
- The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants
De novo assemblers
Performance measures
- ctg/scf/singleton stats
ABYSS
# single reads; one library
# getting the best kmer size
ABYSS -k31 frag_12.fastq -o contigs.fa > ABYSS.log
perl -e 'foreach $k (21,23,25,27,29,31,33,35,37,39,41,43,45,47,49,51) { print "ABYSS -k$k frag_12.fastq -o frag.K$k.fa\n" }'
perl -e 'foreach $k (21,23,25,27,29,31,33,35,37,39,41,43,45,47,49,51) { print "lenseq frag.cor.K$k.fa | getSummary.pl -nh -i 1 -t K$k\n" }' > ABYSS.summary
#paired & single reads abyss-pe -j2 k=31 n=10 name=ecoli lib='lib1 lib2' lib1='lib1_1.fa lib1_2.fa' lib2='lib2_1.fa lib2_2.fa' se='se1.fa se2.fa'
############################## #paired reads; multiple libraries; n: minimum number of links between 2 contigs in a scaffold abyss-pe k=31 n=5 name=genome lib='frag short' frag='frag_1.fastq frag_2.fastq' short='short_1.fastq short_2.fastq' aligner=bowtie >& abyss-pe.log
#paired reads; multiple libraries; n: minimum number of links between 2 contigs in a scaffold abyss-pe k=31 n=5 name=genome lib='frag short' frag=frag_12.cor.fastq short=short_12.cor.fastq aligner=bowtie >& abyss-pe.log
ALLPATHS-LG
The resulting draft genome assemblies have good accuracy, short-range contiguity, long-range connectivity, and coverage of the genome. In particular, the base accuracy is high (≥99.95%) and the scaffold sizes (N50 size = 11.5 Mb for human and 7.2 Mb for mouse) approach those obtained with capillary-based sequencing
Good assemblies definition: ctgLen=~20–100 kb ; scfLen=~10 Mb
Libraries,insert_types* Fragment_size(bp) Read_length(bp) Sequence_coverage(×) Required Protocols Fragment 180 ≥100 45 Yes existing ; try to improve high GC-content reg ShortJump 3,000 ≥100 45 Yes Illumina LongJump 6,000 ≥100 5 No SOLiD EcoP15I based FosmidJump 40,000 ≥26 1 No “ShARC” and “Fosill” developed by BROAD
- Manual
- Data tested on:
- Compile:
#wget ftp://ftp.broadinstitute.org/pub/crd/ALLPATHS/Release-LG/LATEST_VERSION.tar.gz wget ftp://ftp.broadinstitute.org/pub/crd/ALLPATHS/Release-LG/latest_source_code/LATEST_VERSION.tar.gz tar xzvf LATEST_VERSION.tar.gz cd allpathslg-* autoconf ./configure --with-boost=/fs/szdevel/core-cbcb-software/Linux-x86_64/ #ls -l /fs/szdevel/core-cbcb-software/Linux-x86_64/include/boost/ #ll -l /fs/szdevel/core-cbcb-software/Linux-x86_64/lib/libboost_* make –j8 # use ‐j<n> to parallelize compilation
- Compile issues : the static libs (.a) should be linked before the dynamic ones (-l)
Fix: g++ -fopenmp -imacros config.h -Wextra -Wall -Wno-unused -ansi -pedantic -Wno-long-long -Wsign-promo -Woverloaded-virtual \ -Wendif-labels -O3 -ggdb3 -ftemplate-depth-50 -Wno-deprecated -Wno-parentheses -fno-stri ct-aliasing -mieee-fp -iquote . \ -o ErrorCorrectJumpNew -pthread ErrorCorrectJumpNew.o \ -L/fs/szdevel/core-cbcb-software/Linux-x86_64//lib libAllPaths3.a -lboost_system -lboost_filesystem \ -pthread -Wl,-rpath -Wl,/fs/szdevel/core-cbcb-software/Linux-x86_64//lib -o CreateLookupTab -pthread CreateLookupTab.o \ -o SamplePairedReadStatsOld -pthread SamplePairedReadStatsOld.o \ -o SamplePairedReadStats -pthread SamplePairedReadStats.o \ -o ErrorCorrectJump -pthread ErrorCorrectJump.o \
g++ -fopenmp -Wextra -Wall -Wno-unused -Wno-deprecated -ansi -pedantic -Wno-long-long -Wsign-promo -Woverloaded-virtual \ -Wendif-labels -Wno-empty-body -Wno-parentheses -Wno-array-bounds -fno-nonansi-builtins -ftemplate-depth-50 -mieee-fp -fno-strict-aliasing -iquote . -imacros config.h -ggdb3 \ -o ErrorCorrectJumpNew \ -L/fs/szdevel/core-cbcb-software/Linux-x86_64//lib -R/fs/szdevel/core-cbcb-software/Linux-x86_64//lib \ -pthread ErrorCorrectJumpNew.o libAllPathsLG.a -lboost_filesystem
- Configuration:
set path=($path /fs/szdevel/core-cbcb-software/Linux-x86_64/packages/allpaths3/bin/) limit stacksize 100000
- Sequence formatting: sort and add /[12] suffix if needed
Picard java -jar /nfshomes/dpuiu/core-cbcb-software/Linux-x86_64/packages/picard-tools/FastqToSam.jar SAMPLE_NAME=in_12 QUALITY_FORMAT=Standard FASTQ=in_1.fastq FASTQ2=in_2.fastq OUTPUT=out_12.bam java -jar /nfshomes/dpuiu/core-cbcb-software/Linux-x86_64/packages/picard-tools/SamToFastq.jar INPUT=in_12.bam FASTQ=out_1.fastq SECOND_END_FASTQ=out_2.fastq
- in_groups.csv & in_libs.csv files required
echo group_name, library_name, file_name >! in_groups.csv echo frag, frag, chr14_fragment_12.bam >> in_groups.csv echo short, short, chr14_shortjump_12.bam >> in_groups.csv echo long, long, chr14_longjump_12.bam >> in_groups.csv
echo library_name, project_name, organism_name, type, paired, chr14_fragment_size, chr14_fragment_stddev, insert_size, insert_stddev, read_orientation, genomic_start, genomic_end >! in_libs.csv echo frag, genome, genome, fragment, 1, 180, 20, , , inward, 0, 0 >> in_libs.csv echo short, genome, genome, jumping, 1, , , 3000, 300, outward, 20, 81 >> in_libs.csv echo long, genome, genome, jumping, 1, , , 35000, 3500, inward, 20, 56 >> in_libs.csv
- prepare data
BAMsToAllPathsInputs.pl DATA_DIR=$PWD PLOIDY=2 HOSTS=16 GENOME_SIZE=107349540 PICARD_TOOLS_DIR=/nfshomes/dpuiu/core-cbcb-software/Linux-x86_64/packages/picard-tools/ or Allpaths-LG-preprocess.amos
- run assembler
(time RunAllPaths3G PRE=$PWD REFERENCE_NAME=. DATA_SUBDIR=. RUN=allpaths SUBDIR=run ERROR_CORRECTION=True ) >& RunAllPaths3G.log or Allpaths-LG-.amos
- prepare data and run assembler in one subdirectory
~/bin/Allpaths-LG/Allpaths-LG-preprocess.amos -D PLOIDY=1 -D DATA_DIR=$PWD/mydata RunAllPaths3G PRE=$PWD REFERENCE_NAME=. DATA_SUBDIR=mydata RUN=RunAllPaths3G SUBDIR=run >&! mydata.log
- process run log
cat RunAllPaths3G.log | grep RUN | p 'next unless (/^\[/ and $F[1]=~/^\w+/);shift @F; print join " ",@F; print "\n";' | sed -f RUN.sed | p 'print $.*10,": "; print "\$(BINDIR)/" unless(/^ln/); print $_;' > RUN.amos
or
allpathsLogFilter.pl
Example: Ecoli: 0.05 simulated error ; 3 libs 50X,50X,5X
step 2-30,32-36 : read error correction ; should run only once
step program start duration(5+ min)
1 RunAllPaths3G 17:36:59
2 RemoveDodgyReads 17:37:03
3 PreCorrect 17:38:08 9
4 FindErrors 17:47:24 30 !!! most time consuming: 50% of the total time
5 CommonPather 18:27:59 11
6 MakeRcDb 18:38:09
7 Unipather 18:42:05
8 CommonPather 18:43:54
9 MakeRcDb 18:48:50
10 FillFragments 18:50:09
11 CommonPather 18:51:07
12 MakeRcDb 18:54:43
13 Unipather 18:55:08
14 CloseUnipathGaps 18:55:29
15 LittleHelpsBig 18:58:23
16 ShaveUnipathGraph 19:00:19
17 ReplacePairsStats 19:01:44
18 RemoveDodgyReads 19:01:48
19 SamplePairedReadStats 19:02:48
20 UnipathPatcher 19:03:45
21 CommonPather 19:06:42
22 MakeRcDb 19:10:40
23 Unipather 19:10:41
24 FilterPrunedReads 19:10:43
25 CreateLookupTab 19:10:54
26 ErrorCorrectJump 19:11:41
27 SplitUnibases 19:12:26
28 MergeReadSets 19:12:28
29 GenerateLengthsFile 19:12:42
30 MakeRcDb 19:12:52
31 MergeReadSets 19:13:09
32 UnibaseCopyNumber2 19:13:19
33 UnipathLocs3G 19:15:20
34 RemoveDodgyReads 19:15:38
35 ErrorCorrectJump 19:15:46
36 MergeReadSets 19:16:02
37 BuildUnipathLinkGraphs3G 19:16:05
38 SelectSeeds 19:16:23
39 LocalizeReads3G 19:16:36 9
40 MergeNeighborhoods1 19:25:30
41 MergeNeighborhoods2 19:26:48
42 MergeNeighborhoods3 19:26:50
43 DumpHyper 19:27:06
44 RecoverUnipaths 19:27:12
45 FlattenHKP 19:27:54
46 AlignPairsToFasta 19:27:56
47 RemoveHighCNAligns 19:28:12
48 MakeRcDb 19:29:02
49 MakeScaffolds3G 19:29:06
50 PostPatcher 19:29:36
51 FixSomeIndels 19:31:35
52 MakeReadLocs 19:37:53
53 ParseAssemblyReport 19:38:16
- getting the corrected reads
Allpaths-LG-postprocess.amos ... Fastb2Fasta IN=jump_reads_ec.fastb OUT=jump_reads_ec.fasta BREAKCOL=101 ...
Example: S aureus: 0.00 vs 0.02 simulated error ; 4 libs:180_45X,3000_45X,6000_5x,40000_1x
more /fs/szattic-asmg5/dpuiu/HTS/Staphylococcus_aureus/Illumina.sim.180_45X.3000_45X.6000_5x.40000_1x/allpathsCor/README orig(e=0.00;0.02) len allpathsCor(e=0.00;0.02) len:min;max orientation ---------------------------------------------------------------------------------------------------------- frag/1 647052 100 filled_reads_filt.fasta 645445 644487 120 240 inward frag 1294104 100 frag_reads_corr.fasta 1294093 1293557 100 100 inward short 1294104 outward->inward medium 143644 outward->inward long 28728 100 long_jump_reads_ec.fasta 28716 17260* 96 100 inward ---------------------------------------------------------------------------------------------------------- short+medium 1437748 100 jump_reads_ec.fasta 1437188 872734* 97 100 short+medium+long 1466476 100 scaffold_reads.fasta 1465904 889994 96 100 frag+short+medium 2731852 100 reads_merged_ec.fasta 2731281 2166291 97 100 frag+short+medium+long 2760580 100 all_reads.fasta 2178124 1593104 96 240 ----------------------------------------------------------------------------------------------------------
- Notes
On a simulated data set Ploidity = 1 or 2 did not help much the jump outies get reversed
CA
- Version: 6.1
- Web site
- Input: 454, Sanger
- Illumina format (Sanger or Illumina FASTQ)
fastqToCA -libraryname prefix -type sanger -fastq $PWD/prefix.fastq > prefix.frg fastqToCA -insertsize 300 30 -libraryname prefix -type [sanger|illumina] -innie -fastq $PWD/prefix_1.fastq,$PWD/prefix_2.fastq > prefix_12.frg fastqToCA -insertsize 3000 300 -libraryname prefix -type [sanger|illumina] -outie -fastq $PWD/prefix_1.fastq,$PWD/prefix_2.fastq > prefix_12.frg # long inserts
- Command
~/bin/runCA -d . -s runCA.spec -p asm prefix*.frg >& runCA.log
- 454
- must use bog/mer params, otherwise lib insert size get reestimated; portion of scaffolds moved and rotated (compared to the ref seq)
cat runCA.454.spec unitigger = bog ovlOverlapper = mer
- Sanger/Illumina
cat runCA.Sanger.spec unitigger = bog ovlOverlapper = ovl
- Large assemblies, run in parallel
- Repeats: usually end up as degenerates
- Breaks: seem to be caused by short tandem repeats (the multiple contig alignments are adjacent)
CA ShortReads assembly issues
Reads (~100bp) , high error
- GC% bias, nonrandomness & redundancy in shortjump libs
Use estimate genome size runCA genomeSize=...
Redundancy/chimera detection script ~/dpuiu/bin/azimin/filter_library.sh
- Set kmer thold manually to ~10 times the coverage
74X cvg => 750 kmer thold
runCA obtMerThreshold=750 ovlMerThreshold=750
- Recompile the CA code
cat AS_CGW/AS_CGW_dataTypes.h #define CGW_MIN_DISCRIMINATOR_UNIQUE_LENGTH 250
- Avoid unitig breaks caused by fragment libries (~180bp ins)
cat AS_BOG_MateChecker.cc // if (globalStats[lib].samples < 10 ) if (globalStats[lib].samples < 10 || globalStats[lib].stddev < 100 || globalStats[lib].mean < 500)
CA VeryShortReads assembly issues
Reads <40bp , high error
- Recompile the CA code
cat AS_global.h #define AS_READ_MIN_LEN 32 #define AS_OVERLAP_MIN_LEN 30
Read correction
- quake: better
- OBT : takes too long, trims too much seq
Kmers
- decrease kmer size 22->?
Unitigger
- bog
- increase utgErrorRate 0.015->?
- Issues
- Snps in the repetitive unitigs
6 out of 529 unitigs don't align perfectly to genome => misassemblies data : 75bp error-free simulated reads 200bp lib: 25X cvg 6K lib : 14X command: ~/bin/runCA -s ./runCA.spec doOverlapBasedTrimming=0 unitigger=bog utgErrorRate=0.0 cgwErrorRate=0.0 cnsErrorRate=0.0 ovlErrorRate=0.0 -d . -p test test*.frg
show-coords -L 200 test.utg.delta | egrep -n '220002412853|220002412688' 736581 736875 | 1 295 | 295 295 | 99.32 | 4639675 295 | 0.01 100.00 | NC_000913 utg220002412853 [CONTAINS] 736741 737024 | 1 284 | 284 284 | 100.00 | 4639675 284 | 0.01 100.00 | NC_000913 utg220002412688 [CONTAINS]
show-snps test.utg.filter-q.delta | grep 220002412853 736815 A G 235 | 36 61 | 1 0 | 1 1 NC_000913 utg220002412853 736851 A C 271 | 25 25 | 1 0 | 1 1 NC_000913 utg220002412853
Cgw
filter out single fragment unitigs (most of them) and fix aStat from <1 to 1 ~/bin/filterUnitigs.sh
Edena
- Version 2.1.1
- Web site
- Input: Illumina ; unmated only !!!
- Format: seq, fastq
- Command:
edena --readsFile prefix.fastq --prefix prefix --minOverlap [20] # overlap mode edena --edenaFile prefix.ovl -p prefix --overlapCutoff [22] # assembly mode
- Output: no information about the position of the reads in assembly !!!
Newbler
- [2] more info ...
- Input: 454, Sanger
- Format: sff, seq/qual
- The linker is automatically detected only in the sff files
- The linker has to be "manually" removed from seq/qual files
- Q: How to assemble paired reads from seq files?
- A: Header line DIR=F, DIR=R : see "gatekeeper" command
- No way to specify the library insert/std !!!
- The input file order matters !!!
- Can perform slightly better if CA OBT trimmings are used (Ex:Ecoli 454)
- DeNovo commands:
# sff input runAssembly -force -o dir *.sff # seq/qual files ; -p : explicit pair ends test -f prefix.seq test -f prefix.seq.qual runAssembly -o dir [-p] *.seq # step by step; multiple sequence types/insert size; order matters newAssembly . addRun -p Sanger.seq addRun -p 454.flx.sff addRun -p 454.tit.sff runProject .
- RefMapper commands:
#sff input runMapping -o . prefix.1con *.sff # step by step newMapping . addRun -p Sanger.seq addRun -p 454.flx.sff addRun -p 454.tit.sff setRef ref.1con runProject .
- paired reads: must have the same template/library; dir=F|R
Example: >SRR001355.3635.1a template=2020+2021 dir=F library=SRX000348 trim=20-117 ... >SRR001355.3635.1b template=2020+2021 dir=R library=SRX000348 trim=1-130 ...
- config file
assembly/454AssemblyProject.xml Not tested: edit the params in this file and relaunch the assembly
- viewer
toAmos -ace 454Contigs.ace -m prefix.mates -o prefix.afg bank-transact -c -z -b prefix.bnk -m prefix.afg
SGA
- Website
- SGA (String Graph Assembler) is a de novo assembler for DNA sequence reads. It is based on Gene Myers' string graph formulation of assembly and uses the FM-index/Burrows-Wheeler transform to efficiently find overlaps between sequence reads.
- Paper Efficient construction of an assembly string graph using the FM-index : Bioinformatics 2010
- Author website
- Git download
- README
- HELP
- Compile
./configure --with-sparsehash=/fs/szdevel/core-cbcb-software/Linux-x86_64/ --with-bamtools=/fs/szdevel/core-cbcb-software/Linux-x86_64/packages/bamtools/
- Example
#!/bin/tcsh -x setenv THREADS 20 setenv MINQUAL 4 setenv MINOVL 45 setenv MAXERR 0.04 setenv K 31 setenv ITTERATIONS 10 setenv k 27 setenv MINCVG 3 ################################################################## rm frag.pp.* default-* sga preprocess -p 1 -f $MINQUAL frag_?.fastq > frag.pp.fa sga index -t $THREADS frag.pp.fa sga correct -k $K -i $ITTERATIONS -t $THREADS frag.pp.fa -e $MAXERR -m $MINOVL -o frag.pp.ec.fa sga index -t $THREADS frag.pp.ec.fa sga filter -k $k -x $MINCVG frag.pp.ec.fa sga overlap -m $MINOVL -t $THREADS frag.pp.ec.filter.pass.fa sga assemble frag.pp.ec.filter.pass.asqg.gz
SOAPdenovo
- Version: 1.05
- Web site , Manual
- Input: Illumina
- Format: seq, fastq (Sanger/Illumina scale: no conversion neede)
- Other BGI tools for read correction, snp calling, gap closing
Issues
- SOAPdenovo scaff:
- Floating point exception if the insert is too short
- No output difference if the insert size given is longer than the real estimate
Options
soapdenovo all –s config_file –K 25 –R –o graph_prefix
soapdenovo pregraph –s config_file –K 25 [–R -d -p] –o graph_prefix soapdenovo contig –g graph_prefix [–R –M 1 -D] soapdenovo map –s config_file –g graph_prefix [-p] soapdenovo scaff –g graph_prefix [–F -u -G -p]
-s STR configuration file -o STR output graph file prefix -g STR input graph file prefix -K INT K-mer size [default 23, min 13, max63] -p INT multithreads, n threads [default 8] -R use reads to solve tiny repeats [default no] -d INT remove low-frequency K-mers with frequency no larger than [default 0] -D INT remove edges with coverage no larger that [default 1] -M INT strength of merging similar sequences during contiging [default 1, min 0, max 3] -F intra-scaffold gap closure [default no] -u un-mask high coverage contigs before scaffolding [defaut mask] -G INT allowed length difference between estimated and filled gap -L minimum contigs length used for scaffolding
Config file
- Example 1:
max_rd_len=100 #maximal read length ; set to 100 by default; if not changed, longer reads get truncated [LIB] #paired reads avg_ins=200 #average insert size reverse_seq=0 #if sequence needs to be reversed (FF instead of FR : not checked yet) asm_flags=3 #in which part(s) the reads are used; 1:ctg 2:scf 3:ctg+scf 4:gapClosure rank=1 #in which order the reads are used while scaffolding rd_len_cutoff=75 #cut the reads from the current library to this length pair_num_cutoff=3 #cutoff value of pair number for a reliable connection between two contigs or pre-scaffolds. ; 3:shortInserts; 5:longInserts (don't seem to be able to decrease it under 3; larger values help with the repeats) map_len=32 #minimun alignment length between a read and a contig required for a reliable read location. ; 32:shortInserts; 35:longInserts (don't seem to be able to decrease it under 32; larger values help with the repeats) q1=/path/fastq_read_1.fq #fastq file for read fwd; names in q1&q2 don't have to match but the positions do !!! q2=/path/fastq_read_2.fq #fastq file for read rev f1=/path/fasta_read_1.fa #fasta file for read fwd f2=/path/fasta_read_2.fa #fasta file for read rev p=/path/fasta_read_12.fa #fwd followed by rev q=/path/fastq_read.fq #fastq file for read f=/path/fasta_read.fa #fasta file for read [LIB] avg_ins=3000 reverse_seq=1 asm_flags=2 rank=2 pair_num_cutoff=5 map_len=35 q1=... q2=...
iid2uid mapping & posmap file
- Example:
cat s_1.unmated s_1.mates s_2.unmated s_2.mates s_3.unmated s_3.mates ... | p ' print $F[0],"\n"; print $F[1],"\n" if($F[1]);' | nl > s.iid2uid cat s_1.mates s_2.mates s_3.mates ... | p ' print $F[0],"\n"; print $F[1],"\n" if($F[1]);' | nl > s.iid2uid
paste s_1.fastq s_2.fastq | perl -ane ' next unless($.%4==1); 's/\@//g' ; @F=split /\s+/; print $F[0],"\n",$F[1],"\n";' | nl > s.iid2uid sort -nk2 -nk3 s.readOnContig | grep -v ^read | ~/bin/replaceFirstCol.pl -f $1.iid2uid | perl -ane '$F[4]=($F[3] eq "+")?"f":"r"; $F[3]=$F[2]+64; print join "\t",@F; print "\n";' > s.posmap.frgctg
links
- Shows the links between contigs
lenseq test.contig | egrep '1376|2376' ctg len ... 1376 100 2376 69673 ...
egrep '1376|2376' test.scaf #ctg pos dir len #DOWN ctg:totLinks:avgLibMea ... 1376 109421 + 100 #DOWN 2376:52:55 #UP 2334:43:11 2376 109545 + 69673 #DOWN 2380:255:13 #UP 2334:255:8 1376:52:55 ... cat test.links | grep 2376 | grep 1376 #ctg1 ctg2 dist #Links libMea 1376 2376 34 32 240 1376 2376 91 20 6054
avgLibMea= (sum #Links*libMea)/(sum #Links)
Commands
- All at once:
SOAPdenovo all -s SOAPdenovo.config -o prefix # all: do all assembly steps
- Step by step:
1. (time SOAPdenovo pregraph -s SOAPdenovo.config -o prefix -K 23 -p 6 ) >>& SOAPdenovo.log prefix.kmerFreq # up to 256 prefix.edge # FASTA format prefix.preArc prefix.vertex # md5 sums ??? prefix.preGraphBasic
2. (time SOAPdenovo contig -g prefix -M 1 ) >>& SOAPdenovo.log prefix.contig prefix.ContigIndex prefix.updated.edge prefix.Arc
3. (time SOAPdenovo map -s SOAPdenovo.config -g prefix -p 6 ) >>& SOAPdenovo.log prefix.readOnContig prefix.peGrads prefix.readInGap
4. (time SOAPdenovo scaff -g prefix -F ) >>& SOAPdenovo.log prefix.newContigIndex prefix.links prefix.scaf prefix.scaf_gap prefix.scafSeq prefix.gapSeq
Run time
- On Saureus simulated data set
1. 216.460u 2.792s 0:52.27 419.4% 2. 0.425u 0.011s 0:00.50 86.0% 3. 59.921u 1.597s 0:21.21 290.0% 4. 6.113u 0.263s 0:06.22 102.4%
- Larger genomes ???
Output
- No information about the position of the reads in assembly !!!
cat scaffold1.filter-q.delta | ~/bin/delta2posmap.pl | sort -nk3 | head -20 | pretty
Adding more reads for scaffolding
Files to link:
perl -e '@F=split /\s+/,"Arc contig ContigIndex edge kmerFreq peGrads preArc preGraphBasic vertex"; \
foreach (@F) { print "ln -s ../SOAP-prev/*.$_\n" }'
GapCloser (works with SOAPdenovo)
- Version: 1.10
- Web site
~/szdevel/bin/SOAPGapCloser -b SOAPdenovo.config -a Bb.scafSeq -o Bb.scafSeq.new -t 12 => Bb.scafSeq.new : FASTA file(Bb.scafSeq with fewer N's) Bb.scafSeq.new.fill : gap fill status Example: >scaffold2 175.2 ... #begin end 5'fill 3'fill closed? 55203 57185 0 23 0 69718 71607 0 28 0 79195 79219 24 0 1 80681 80741 0 23 0 81738 81876 0 37 0 ...
GC vs Coverage correlation
infoseq -description asm.K31.contig | p '/cvg_(\d+.\d+)/; print "$F[0] $F[1] $F[2] $1\n" ;' >! asm.K31.contig.infoseq
cat asm.K31.contig.infoseq | ~/bin/getCorrelationDescriptive.pl -i 2 -j 3 -min 100 cat asm.K31.contig.infoseq | ~/bin/getCorrelationDescriptive.pl -i 2 -j 3 -min 1000 ...
SOAPdenovo-prepare
- Data Preparation Module generates necessary data for SOAPdenovo to run "map" and "scaff" steps from Contigs generated by SOAPdenovo or other assemblers with various length of kmer.
- options:
* -g [string] Prefix of output. * -K [int] Kmer length. * -c [string] Input Contigs FASTA. (Filename cannot be prefix.contig)
- Example:
/fs/szdevel/core-cbcb-software/Linux-x86_64/bin/SOAPdenovo-prepare -K 31 -c default-contigs.fa -g genome => genome.ContigIndex genome.updated.edge genome.contig genome.Arc genome.conver genome.preGraphBasic
SOAPdenovo map -s SOAPdenovo.config -g genome -p 20 SOAPdenovo scaff -g genome -F -p 20
- Eaxmple: use it if the jumpreads are < contigging kmer size
more ~/bin/SOAPdenovo-63mer.sh #!/bin/sh -x # Usage: SOAPdenovo-63mer.sh prefix ctgK scfK # pregraphing ... SOAPdenovo-63mer pregraph -s SOAPdenovo.config -K $2 -p 16 -o $1 >! SOAPdenovo.log # contigging ... SOAPdenovo-63mer contig -g $1 -M 1 >> SOAPdenovo.log # preparing ... mv $1.contig $1.contig.tmp SOAPdenovo-prepare -K $3 -c $1.contig.tmp -g $1 >> SOAPdenovo.log # read mapping ... SOAPdenovo-63mer map -s SOAPdenovo.config -g $1 -p 16 >> SOAPdenovo.log # scaffolding ... SOAPdenovo-63mer scaff -g $1 -F -p 16 >> SOAPdenovo.log
Velvet
Concerning read orientation, Velvet expects paired-end reads to come from opposite strands facing each other, as in the traditional Sanger format. If you have paired-end reads produced from circularisation (i.e. from the same strand), it will be necessary to replace the first read in each pair by its reverse complement before running velveth.
- Compile:
make ’CATEGORIES=16’ \ # max libraries
’MAXKMERLENGTH=64’ \
’OPENMP=1’ \ # multithread
’BIGASSEMBLY=1’ \ # more than 2.2 billion reads
’LONGSEQUENCES=1’ # contigs longer than 32kb long
make CATEGORIES=16 MAXKMERLENGTH=64 OPENMP=1 BIGASSEMBLY=1 LONGSEQUENCES=1
- Input: 454 , Illumina, ABI SOLiD as well? , AssembledSequences
- Format: seq, fastq (Sanger/Illumina style no conversion needed) all mates should be innies
- 454:
- does poorly
- slightly better if the cvg is given
- same results for fasta/fastq input
- results get worse for higher cvg
- Commands:
# unmated reads velveth . <hash_size> -fastq -short *.fastq velvetg . -amos_file yes -read_trkg yes -exp_cov <cvg> # mated reads shuffleSequences_fastq.pl prefix_1.fastq prefix_2.fastq prefix_12.fastq velveth . <hash_size> -fasta -short prefix.fastq -shortPaired prefix_12.fastq velvetg . -ins_length <mean> -ins_length_sd <std> -amos_file yes -read_trkg yes -exp_cov <cvg>
#reference based bowtie prefix.1con -1 prefix_1.fastq -2 prefix_2.fastq -l 28 -n 2 --best -I 100 -X 200 -p 12 --sam | sort > prefix.bowtie.sam velveth . 21 -reference prefix.1con -shortPaired -sam prefix.bowtie.sam velvetg . -exp_cov auto -ins_length 150 -scaffolding yes -exportFiltered yes -unused_reads yes
Aleksey's create_k_unitigs
- Usage
- genome8.umd.edu
jellyfish count -m 31 -s 1073741824 -r -C -t 16 -o frag.k31.jf *.fa mv -i frag.k31.jf_0 frag.k31.jf create_k_unitigs -C -l 64 -m 5 -M 5 -t 16 -o frag.k31 frag.k31.jf cat frag.k31_*.fa > frag.k31.fa cat frag.k31_*.counts > frag.k31.counts rm frag.k31_* ~alekseyz/myprogs/getNumBasesPerReadInFastaFile.perl frag.k31.fa | sort -gr | head
- cbcb
/fs/szdevel/dpuiu/filter_illumina_data_quals.pl /fs/szdevel/dpuiu/k_unitig-0.0.1/bin/
cat *fastq | /fs/szdevel/dpuiu/filter_illumina_data_quals.pl '#' > genome.seq /fs/szdevel/dpuiu/k_unitig-0.0.1/bin/jellyfish count -s 200000000 -m 31 -t 16 -r -C genome.seq /fs/szdevel/dpuiu/k_unitig-0.0.1/bin/error_correct_reads -d mer_counts.hash_0 -t 16 -m 3 frag_1.fastq
Jellyfish
jellyfish count -m 18 -s 1073741824 --both-strands -t 16 -o frag.k18.jf frag_?.*fastq jellyfish merge frag.k18.jf_* -o frag.k18.jf rm frag.k18.jf_*
jellyfish histo frag.k18.jf > frag.k18.histo
jellyfish stats frag.k18.jf > frag.k18.stats
jellyfish stats --f frag.k18.jf > frag.k18.fasta
jellyfish stats --c frag.k18.jf | awk '{print $2,$1}' > frag.k18.tab
Scaffolding
Bambus
Comparative assemblers
Performance measures:
- ctg/scf/singleton stats
- a related finished genome(s)must be available
- Q:which is the closest relative? A:align the reads to the refs and identify the one with the max number of hits
- Q:how many repeats in the ref & how well are they assembled
- Q: reference coverage: how many gaps are left?
- comparative assembly to a very close/more distant relative
AMOScmp-shortReads
- Web site
- Version:
- Input: 454, Illumina
- Format: amos
- 454 trimming:
- alignment based trimming => longer/fewer contigs
- OBT trimming => shorter/more contigs, fewer snps & breaks
- lucy quality based trimming does not help much
- Illumina trimming
- Trim n bases at the 3'
- Trim all bases with quality < n at the 3' & 5'
- Delete all reads which contain N's
- Commands:
AMOScmp-shortReads prefix # 454 : use nucmer aligner AMOScmp-shortReads-alignmentTrimmed prefix # 454 : use nucmer aligner AMOScmp-shortReads -D DOSOAP=1 prefix # Solexa : use soap aligner
- Can use nucmer/soap for read alignment
nucmer --maxmatch -l 16 -c 32 soap -p 2 -r 2 -v 3 -g 3 # p: parallel # r:2 report multiple occurances # v: max mismatch # g: max gap len
- Breaks
- Q: When the assembly is aligned back to the ref => how many breaks,snps ?
- If several reads map partially to 2 different locations withing few hundred bp of one another & the CONTAINED reads in between align to multiple regions => excision of the repeat
- by decreasing the "casm-layout -g" param from 1000 to 20(?) we can solve some of the problems
Maq
- Version: 0.7.1
- Web site
- Heng Li's presentation
- bioinfo.genopole-toulouse.prd.fr
- Maqview
- BreakDancer-0.0.1 is a Perl package that provides genome-wide detection of structural variants from next generation paired-end sequencing reads.
- Input: Illumina
- Format: fastq
- Commands:
maq.pl easyrun prefix.1con prefix.fastq # single reads maq.pl easyrun prefix.1con prefix.fwd.fastq prefix.rev.fastq -p -a [250] # paired reads; max insert size defaults to 250bp ; NOT WORKING CORRECTLY !!! (0 paired reads reported)
- Performs very well for close ref (>99% id); not that well for further ref (<98% id)
Newbler
- See prev section
- Commands:
# reference based runMapping -o . prefix.1con *.sff
Other assemblers
ABBA
minimus2
#!/bin/tcsh ln -s ../ cat ref.seq qry.seq > prefix.seq toAmos -s prefix.seq -o prefix.afg set REFCOUNT=`grep -c ">" ref.seq` minimus2 -D REFCOUNT=$REFCOUNT -D MINID=94 -D OVERLAP=20 prefix
minimusN
cat ref.asm/ref.scf.fasta | ~/bin/assignFastaIds.pl -p ref | tee ref.scf.fasta | ~/bin/scafffasta2fasta.pl > ref.ctg.fasta cat qry.asm/qry.scf.fasta | ~/bin/assignFastaIds.pl -p qry | tee qry.scf.fasta | ~/bin/scafffasta2fasta.pl > qry.ctg.fasta ~/bin/nucmer.amos -D REFN=18000 -D REF=ref.ctg.fasta -D SEQS=qry.ctg.fasta -D MINMATCH=20 -D MINCLUSTER=40 -D BREAKLEN=20 -D MAXGAP=20 ref-qry delta-filter -i 94 genome.filter-1.delta | /nfshomes/dpuiu/szdevel/SourceForge/AMOS/bin/delta2tab.pl | sort -nk1 -nk6 -nk7 ~/bin/AMOS/preMinimus2 # to get qry breaks minimusN -D REFCOUNT=$REFCOUNT
Sequence aligners
Bowtie
- No gapped alignments (yet)
-n : max mismatches in seed (can be 0-3, default: -n 2) (Soap??) -l : seed length for -n (default: 28) --best : hits guaranteed best stratum; ties broken by quality --strata : hits in sub-optimal strata aren't reported (requires --best)
- Create an index
bowtie-build ref.fasta ref.fasta
- Aligned mated & unmated reads
Example: # qry_1.fastq & qry_2.fastq : mated reads # qry_0.fastq : unmated reads bowtie ref.fasta -1 qry_1.fastq -2 qry_2.fastq -l 28 -n 2 -I 6000 -X 10000 --best -p 8 > qry_12.bowtie bowtie ref.fasta qry_0.fastq -l 28 -n 2 --best -p 8 > qry_0.bowtie
- --chunkmbs <int> max megabytes of RAM for best-first search frames (def: 64)
increase it to 512 or 1024 in case of error
Maq / Bwa
- bwa
- index reference
bwa index ref.fasta
- short single reads
bwa aln ref.fasta qry.fastq > qry.sai bwa samse ref.fasta qry.sai qry.fastq > qry.sam
- short paired reads
bwa aln ref.fasta qry_1.fastq > qry_1.sai bwa aln ref.fasta qry_2.fastq > qry_2.sai bwa sampe ref.fasta qry_1.sai qry_2.sai qry_1.fastq qry_2.fq > qry.sam
- long reads
bwa bwasw ref.fasta qry.fastq > qry.sam
- Example
bwa bwasw cChloroplast.fa test.fastq | sed 's/555//g' | pretty @SQ SN:cChloroplast LN:120481 match.fwd 0 cChloroplast 1 18 100M * 0 0 TTATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGTTGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGATTA ... AS:i:100 XS:i:86 XF:i:2 XE:i:3 XN:i:0 5.fwd 0 cChloroplast 3 36 2S98M * 0 0 NNATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGTTGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGATTA ... AS:i:98 XS:i:84 XF:i:3 XE:i:2 XN:i:0 3.fwd 0 cChloroplast 1 15 98M2S * 0 0 TTATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGTTGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGATNN ... AS:i:98 XS:i:86 XF:i:2 XE:i:3 XN:i:0 5.rev 16 cChloroplast 2 36 1S99M * 0 0 NNATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGTTGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGATTA ... AS:i:99 XS:i:85 XF:i:3 XE:i:2 XN:i:0 3.rev 16 cChloroplast 1 15 98M2S * 0 0 TTATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGTTGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGATNN ... AS:i:98 XS:i:86 XF:i:2 XE:i:3 XN:i:0 ins.fwd 0 cChloroplast 3 40 2S48M2I50M * 0 0 TAATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGTNNTGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGATTA ... AS:i:89 XS:i:75 XF:i:3 XE:i:2 XN:i:0 del.fwd 0 cChloroplast 3 41 2S47M2D49M * 0 0 TAATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGATTA ... AS:i:87 XS:i:73 XF:i:3 XE:i:2 XN:i:0
bwa bwasw -H cChloroplast.fa test.fastq | pretty @SQ SN:cChloroplast LN:120481 match.fwd 0 cChloroplast 1 18 100M * 0 0 TTATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGTTGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGATTA ... AS:i:100 XS:i:86 XF:i:2 XE:i:3 XN:i:0 5.fwd 0 cChloroplast 3 36 2H98M * 0 0 ATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGTTGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGATTA ... AS:i:98 XS:i:84 XF:i:3 XE:i:2 XN:i:0 3.fwd 0 cChloroplast 1 15 98M2H * 0 0 TTATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGTTGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGAT ... AS:i:98 XS:i:86 XF:i:2 XE:i:3 XN:i:0 5.rev 16 cChloroplast 2 36 1H99M * 0 0 NATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGTTGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGATTA ... AS:i:99 XS:i:85 XF:i:3 XE:i:2 XN:i:0 3.rev 16 cChloroplast 1 15 98M2H * 0 0 TTATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGTTGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGAT ... AS:i:98 XS:i:86 XF:i:2 XE:i:3 XN:i:0 ins.fwd 0 cChloroplast 3 40 2H48M2I50M * 0 0 ATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGTNNTGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGATTA ... AS:i:89 XS:i:75 XF:i:3 XE:i:2 XN:i:0 del.fwd 0 cChloroplast 3 41 2H47M2D49M * 0 0 ATCCACCTATTGAAATAGATTCAACAGCGGCTAGATCCAGAGGAAAGGTGAGCATTACGTTCGTGCATAACTTCCATACCTAGGTTAGCACGATTA ... AS:i:87 XS:i:73 XF:i:3 XE:i:2 XN:i:0
bwa bwasw -H cChloroplast.fa test.fastq | ~/bin/AMOS/sam2posmap.pl | pretty @SQ SN:cChloroplast LN:120481 match.fwd cChloroplast 0 100 f 0 100 100 100.00 5. fwd cChloroplast 2 100 f 2 100 98 100.00 3.fwd cChloroplast 0 98 f 0 98 98 100.00 5.rev cChloroplast 1 100 r 1 100 99 100.00 3.rev cChloroplast 0 98 r 0 98 98 100.00 ins.fwd cChloroplast 2 102 f 2 100 98 97.96 del.fwd cChloroplast 2 98 f 2 100 98 97.96
Nucmer
- Run on parallel on big data sets
#!/bin/tcsh set REFN=`grep -c ">" ref.fasta` @ REFN/=20 /nfshomes/dpuiu/bin/nucmer.amos -D REFSEQ=$PWD/ref.fasta -D QRYSEQ=$PWD/qry.fasta -D REFN=$REFN ref-qry
SOAP2
- No gapped alignments
- Create an index
2bwt-builder ref.fasta
- Align mated & unmated reads
-l : alignment seed -s : minimal alignment length -n : filter low-quality reads containing >n Ns before alignment, [5] -r : how to report repeat hits, 0=none; 1=random one; 2=all, [1] -v : maximum number of mismatches allowed on a read. [5] bp -g : one continuous gap size allowed on a read. [0] bp
Example: don't forget "-2" soap2 -D ref.index -a qry_1.fastq -b qry_2.fastq -o qry_12.mated.soap2 -2 qry_12.single.soap2 -l 28 -v 2 -n 0 -m 6000 -x 10000 -p 8 soap2 -D ref.index -a qry_0.fastq -o qry_0.soap2 -l 28 -v 2 -n 0 -p 8
Alignment processing
Samtools
http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit http://www.broadinstitute.org/gsa/wiki/index.php/Converting_Illumina_output_to_BAM_files
Data sets
Sources
- NCBI SRA
- SRA to close
- ENA European Nucleotide Archive (?)
Escherichia coli str. K-12 substr. MG1655
- SRA Study E coli Whole Genome Sequencing on 454 and Illumina (454 unpaired & Illumina reads)
- SRA Study 454 sequencing of Escherichia coli str. K-12 substr. MG1655 genomic paired-end library (454 paired reads)
- 29 complete strains: NCBI search: "Escherichia coli"[Organism] AND pt_default[prop] AND seqstat_complete[PROP]
- /fs/szdata//ncbi/genomes/Bacteria/Escherichia_coli_*/
- chromosome lengths 4.5-5.5Mbp
- plasmids?
- Ref (same species):
len gc% NC_000913 4639675 50.79 Escherichia coli str. K-12 substr. MG1655, complete genome (same substr?) 2.9%repeats NC_010468 4746218 50.87 Escherichia coli ATCC 8739, complete genome (99% id) NC_008253 4938920 50.52 Escherichia coli 536, complete genome (97% id)
- Ref (same genus):
NC_011740 4588711 49.94 Escherichia fergusonii ATCC 35469, complete genome (94% id)
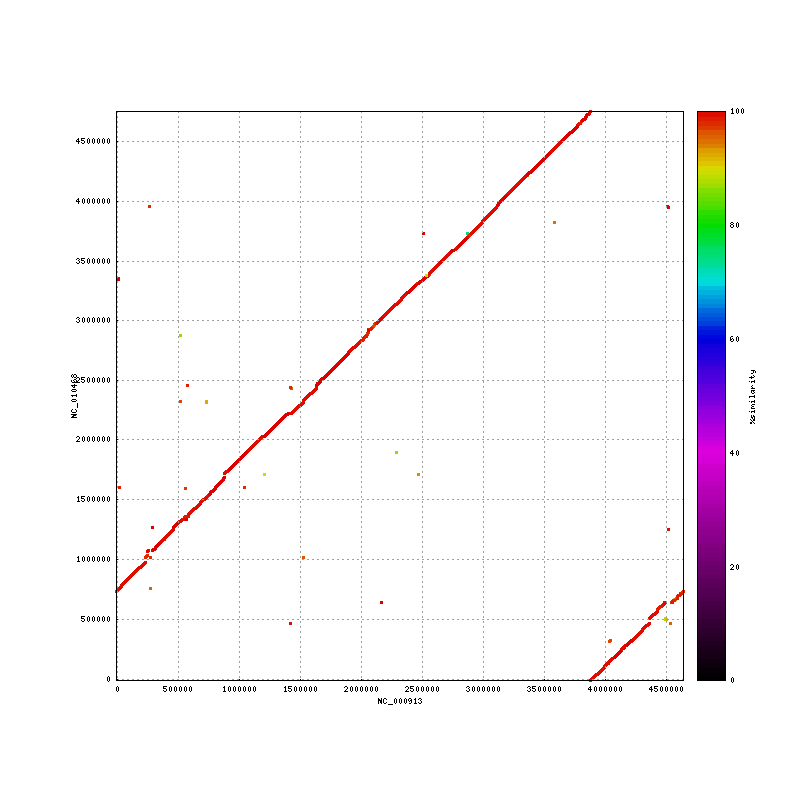
- Ecoli K12 vs ATCC alignment : 99% identity in aligned regions ; 0.926% identity overall Media:Ecoli.K12-ATCC.png
{kind=link}
. elem min q1 q2 q3 max mean n50 sum 0cvg 130 6 153 756 2085 18927 2307 7560 299947 (6.4% of the K12 genome not present in ATCC) %id 4929904 68.73 98.95 99.21 99.38 100.00 98.52 99 .
- Ecoli K12 vs 536 alignment : 97% identity => more distant
. elem min q1 q2 q3 max mean n50 sum 0cvg 375 2 134 461 1549 30645 1854.35 7094 695383 (14.98% of the K12 genome not present in 536) %id 4143854 79.11 96.81 97.48 97.88 100.00 97.17 97 .
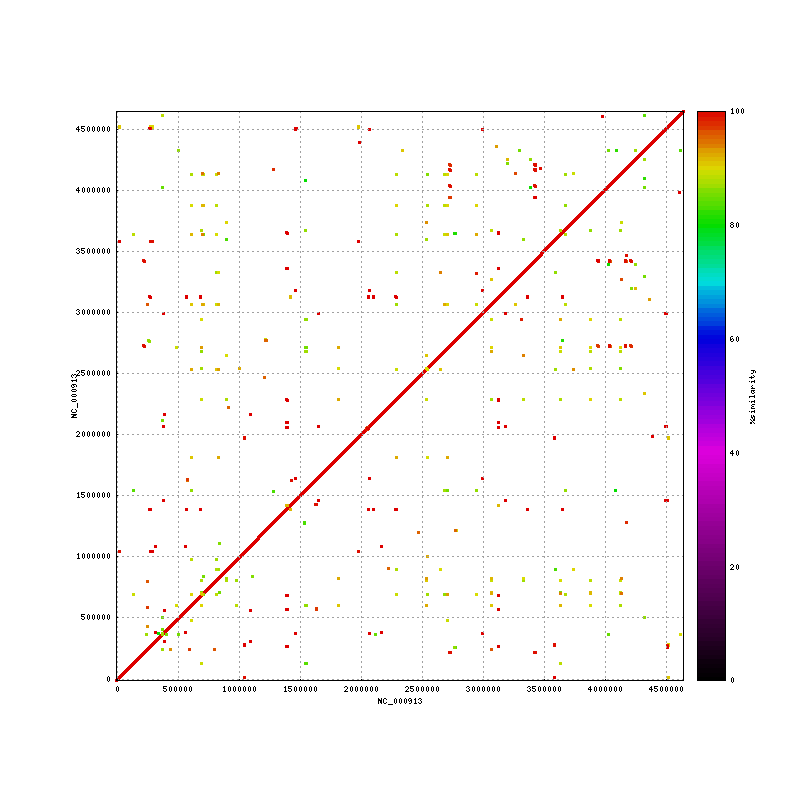
- Ecoli K12 Repeats/Uniq regions (repeat-match based) : max uniq region is 138Kbp (the "best" we can do with short unmated reads) Media:Ecoli.K12-K12.png
{kind=link}
. elem min q1 q2 q3 max mean n50 sum repeats.36bp+ 701 36 42 57 132 3903 197.46 848 138420(2.9%) repeats.100bp+ 202 100 155 271 756 3903 550 1207 111011(2.4%) repeats.200bp+ 126 200 290 541 1208 3903 794.88 1221 100155(2.1%) uniq 646 1 10 84 8366 138200* 6970.60 28028 4503010
- Longest repeat: 3903b : 3 complete + 8 partial(33%+)
223979 227889 | 3903 2 | 3911 3902 | 99.23 | 4639675 3903 | 0.08 99.97 | NC_000913 NC_000913.3422674-3426576 [CONTAINS] 2725076 2727319 | 2 2245 | 2244 2244 | 99.60 | 4639675 3903 | 0.05 57.49 | NC_000913 NC_000913.3422674-3426576 2727589 2728971 | 2521 3903 | 1383 1383 | 100.00 | 4639675 3903 | 0.03 35.43 | NC_000913 NC_000913.3422674-3426576 3422674 3426576 | 1 3903 | 3903 3903 | 100.00 | 4639675 3903 | 0.08 100.00 | NC_000913 NC_000913.3422674-3426576 [CONTAINS] 3940039 3941416 | 3903 2526 | 1378 1378 | 99.64 | 4639675 3903 | 0.03 35.31 | NC_000913 NC_000913.3422674-3426576 3941605 3943857 | 2245 2 | 2253 2244 | 99.33 | 4639675 3903 | 0.05 57.49 | NC_000913 NC_000913.3422674-3426576 4033762 4037673 | 3903 2 | 3912 3902 | 99.41 | 4639675 3903 | 0.08 99.97 | NC_000913 NC_000913.3422674-3426576 [CONTAINS] 4164890 4166276 | 3903 2518 | 1387 1386 | 99.57 | 4639675 3903 | 0.03 35.51 | NC_000913 NC_000913.3422674-3426576 4166542 4168794 | 2245 2 | 2253 2244 | 99.11 | 4639675 3903 | 0.05 57.49 | NC_000913 NC_000913.3422674-3426576 4206378 4207764 | 3903 2518 | 1387 1386 | 99.57 | 4639675 3903 | 0.03 35.51 | NC_000913 NC_000913.3422674-3426576 4207944 4210196 | 2245 2 | 2253 2244 | 99.20 | 4639675 3903 | 0.05 57.49 | NC_000913 NC_000913.3422674-3426576
- Ecoli K12 Repeats (NCBI annotation)
. elem min q1 q2 q3 max mean n50 sum repeats 399 14 32 82 111 1443 191.69 980 76485 # much fewer than the ones I found genes 4485 14 465 813 1218 7077 922.17 1188 4135942 # ~ 62 genes (42Kb) CONTAINED by 42 annotated repeats
- Ecoli K12 kmers
23mers: most frequent occurance is 77 31mers: 46 41mers: 11
- Ecoli K12 unique regions (Example)
1745081-1952189
# ~ 132 genes (75K) CONTAINED by 79 repeat-match repeats
- Location on disk:
/fs/szattic-asmg4/dpuiu/HTS/Escherichia_coli/
Read stats
- 454 SRA
. elem min max mean n50 sum cvg mea;std q20Pos SRA accession(s) 454 257,477 52 943 262 263 67640102 14.57 . SRX012358,SRX012359 454 paired 322,386 4 831 185 248 59677025 12.86 3500 SRX000348 (different study,only run)
- Illumina SRA
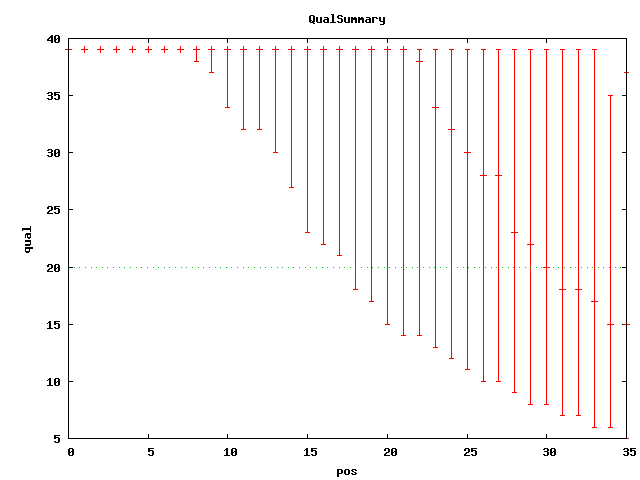
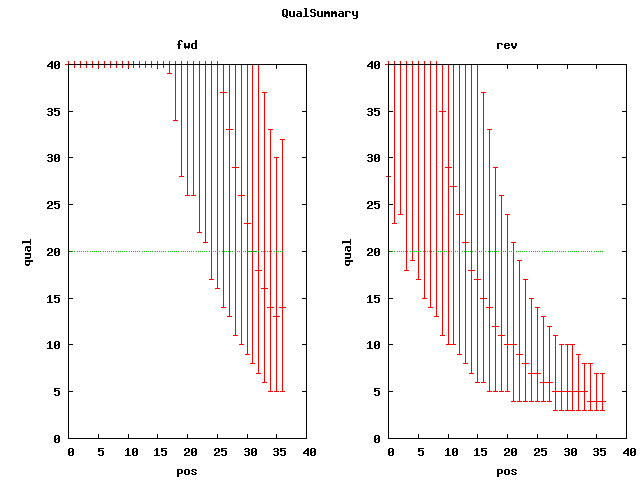
. elem min max mean n50 sum cvg mea;std q20Pos SRA accession(s) Illumina 6,725,378 36 36 36 36 242113608 52.18 . 30 SRX007752 qual plot Illumina paired(37) 9,928,004 37 37 37 37 367336148 79.17 178 30;13 SRX012850 qual plot Illumina paired(101) 20,635,060 101 101 101 101 . 449 168 50;13 SRX016044 SRR034509 qual plot # based on alignments to ref, mea=~64
{kind=link}
{kind=link}
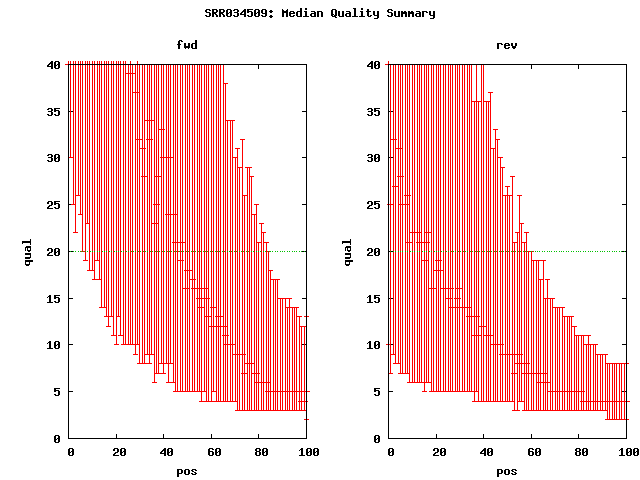{kind=link}
Illumina paired(101) ? 101 101 101 101 . 449 150 50;13 ERX008638 Illumina paired(36) ? 500 SRX000430 Illumina paired(36) ? 200 SRX000429
- Illumina company: data described in Illumina Technical Notes; complete dataset received from Amy Bouck-Knight(techsupport@illumina.com)
. elem min max mean n50 sum cvg mea;std q20Pos min;max Illumina paired(75) 20,304,466 75 75 75 75 1522834950 328 210;10 75;75 180;240 # Oct2010 *0.15 => 50X # s_6 ; 53% of reads are error free Illumina paired(35) 6,211,692 35 35 35 35 217409220 46 6054:280 35;35 5214;6894 # Oct2010 *0.60 => 28X # s_7 ; 78% of reads are error free Illumina paired(35) 4,851,382 35 35 35 35 169798370 36 8917;700 35;35 6817;11017 # Oct2010 *0.77 => 28X # s_4 ; 78% of reads are error free
- Illumina simulated
. elem min max mean n50 sum cvg mea;std q20Pos Illumina paired(75) 210 Illumina paired(75) 6054
Read alignments
# ref: Ecoli K12
total aligned aligned% program
454 257477 257372 99.96 nucmer -l 16 -c 32
454 paired 322386 286115 88.75 nucmer -l 16 -c 32
Illumina 6725378 4109521 61.10 soap -g 5 -v 5
Illumina paired 9928004 3819582(3663441 fwd+156141 rev) 38.47 soap -g 5 -v 5
454 (14.57X read cvg)
- DeNovo assembly
# ctg stats
ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks comments
CA.bog** 176 1005 6116 17855 37563 169694 25899 44290 4558385 257477 1145 59607 1003 12 only contigs used for alignments; if deg used 0cvgSum=32455
newbler.deNovo 274 100 1738 8645 25332 125168 16651 35805 4562444 257477 20255 68423 1242 5
velvet 41859 45 54 67 96 9132 156 404 6566010(?) 257477 802 200706 45017 37 most uncovered regions
- DeNovo merge assembly (minimus2; singletons were merged in the contigs file)
# ctg stats
ctgs+sing min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks comments
CA.bog-newbler.deNovo 135 100 2036 19279 52988 224358 35628 79466 4809817 450 0 47585 1011 15
- Reference based assembly (Ecoli K12)
# ctg stats
ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks comments
AMOScmp.alignmentTrimmed** 8 721 178178 548382 1208791 1574740 579935 1208791 4639482 257477 11472 48 1155 0 nucmer alignment trimmed reads
AMOScmp.orig 269 243 4062 10640 23598 146792 17230 31488 4635098 257477 59369 9508 3338 1 original reads
AMOScmp.lucy 181 256 8180 17089 34961 161650 25608 40906 4635189 257477 45663 5839 2393 1 Lucy quality trimmed reads
AMOScmp.OBT 59 386 25713 51940 127336 280148 78607 135820 4637865 257477 723 1524 926 0 OBT trimmed reads
newbler.refMapper 70 196 770 33983 88976 327077 65412 178569 4578847 257477 143 25805 1985 0 most uncovered regions ???
- Reference based K12 & deNovo merge (minimus2; singletons were merged in the contigs file) : not much help
# ctg stats
ctgs+sing min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks comments
CA.bog-AMOScmp.at.K12 24 694 27931 52358 62326 2123245 218921 1554570 5254104 184 0 122 1346 13
- Reference based assembly stats (Ecoli ATCC)
ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks comments AMOScmp.alignmentTrimmed* 199 34 695 5745 26504 292814 21784 75690 4335019 257477 28327 293438 2304 23 breaks are adjacent (tandem repeats?) AMOScmp.OBT 314 77 914 6225 16704 166680 13756 37619 4319673 257477 20556 307393 1645 8 breaks are adjacent (tandem repeats?) newbler.refMapper 213 105 615 6653 25594 211153 19987 57867 4257433 257477 15725 333122 2385 46 breaks are adjacent (tandem repeats?)
- Reference based assembly (Ecoli 536)
# ctg stats
ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks comments
AMOScmp.alignmentTrimmed 601 77 1080 3820 8977 61585 6525 12044 3921648 257477 46572 701339 1406 5
newbler.refMapper 476 101 747 4112 10299 97951 8184 18241 3895796 257477 35796 729086 3004 77
- Read trimming
. elem min q1 q2 q3 max mean n50 sum CA.ORIG,CLR 252939 68 252 261 272 823 263.10 263 66547020 CA.OBTINITIAL 252939 67 250 260 271 724 260.52 261 65894538 CA.OBTCHIMERA,LATEST 252939 64 220 244 257 610 231.39 247 58528503 AMOScmp.K12 257372 33 241 252 263 342 246.44 254 63425475 AMOScmp.ATCC 242665 32 239 252 262 337 244.32 253 59286793 AMOScmp.536 223903 32 237 250 261 342 241.07 252 53977199
- Repeat stats
elem min q1 q2 q3 max mean n50 sum genome 701 36 42 57 132 3903 197.46 848 138420 CA.bog 516 36 40 49 78 848 72.84 79 37585 # may repeats end up in degenerates newbler.deNovo* 576 36 41 52 88 1409 106.82 162 61528 # "long" repeats have only 1 copy in the assembly velvet 123 36 39 43 56 128 51.82 48 6374 AMOScmp.alignmentTrimmed.K12* 680 36 42 59 133 3903 200.92 855 136627 AMOScmp.OBT.K12 675 36 42 58 132 3903 201.00 926 135676 newbler.refMapper.K12 598 36 41 52 88 569 82.42 95 49288 # surprisingly "bad" for a comparative assembler AMOScmp.alignmentTrimmed.ATCC 601 36 42 58 121 3903 179.94 575 108146 AMOScmp.OBT.ATCC 583 36 42 58 123 3903 182.45 581 106370 newbler.refMapper.ATCC 536 36 41 53 95 1208 106.75 156 57217
- Comments:
- all repeats are CONTAINED
- Many gaps match the repeats
- Ref based assemblies to Ecoli ATCC & 536 contain many breaks
- velvet assembly very fragmented; many regions covered by multiple contigs
- Viewers
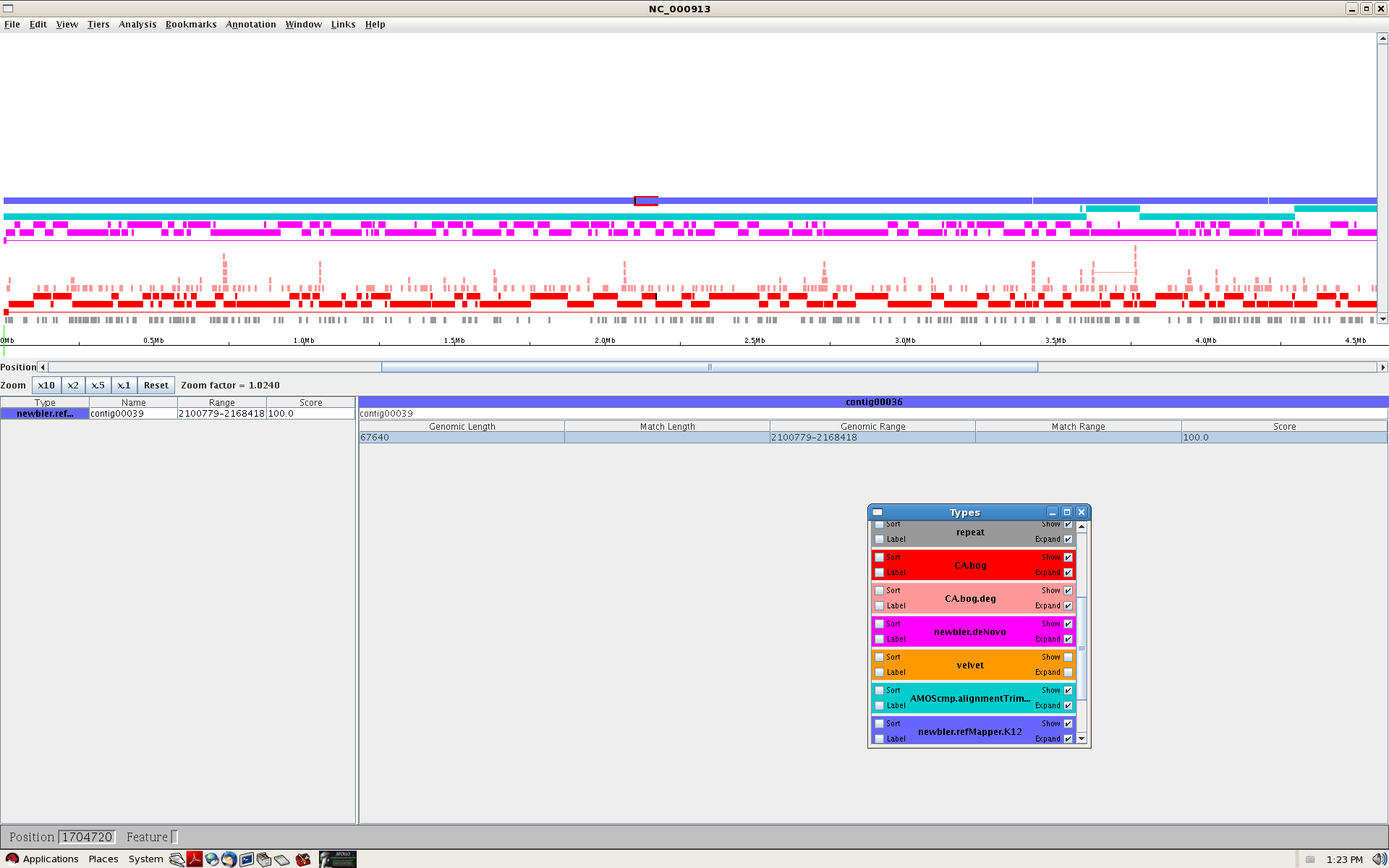{kind=link}
454 paired (12.86X cvg) on FASTQ files
- DeNovo assembly
# contig stats
ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks comments
CA.bog* 351 96 4154 9544 16800 96947 13097 21709 4597092 322386 4318 30577 1227 21 # error in the orig .frg files (FWD-FWD SRA lib => outie mates)
newbler.deNovo 801 100 1402 3701 7530 57354 5645 9907 4522066 322386 1094 86176 640 4
# scaffold stats
scf min q1 q2 q3 max mean n50 sum
CA.bog* 15 292 90673 198619 447960 882752 308205 707016 4623076
newbler.deNovo 23 2062 2637 100587 210519 1824862 203995 598401 4691889
454 vs 454 paired CA.bog assembly qc
- Reference based assembly(Ecoli K12)
# contig stats
ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks comments
AMOScmp.alignmentTrimmed* 94 106 10818 31335 80022 303035 49345 90573 4638465 322386 40727 2552 848 2
AMOScmp 506 53 2633 6051 12524 61278 9156 16014 4633422
newbler.refMapper 114 100 7026 31322 55148 203278 40126 80905 4574379 # error: annotation missing from FASTA headers
newbler.refMapper.redo 801 100 1402 3701 7530 57354 5645 9907 4522066 322382 1094 86176 633 4 # much worse than newbler.refMapper ; 51% mates same ctg, 36% link , 11% FalsePair !!!
- Repeat stats
elem min q1 q2 q3 max mean n50 sum
genome 701 36 42 57 132 3903 197.46 848 138420
CA.bog.redo* 643 36 42 56 140 3903 202.61 926 130277 !!! longest repeat(3903 bp) : 1 out of 3 copies present in assembly;
2nd longest repeat(3240 bp) : 6 out of 7 copies present in assembly;
newbler.deNovo.redo 534 36 41 51 85 1811 116.04 245 61965
velvet 194 36 39 47 69 848 67.57 69 13108
AMOScmp.alignmentTrimmed.K12* 681 36 42 59 132 3903 200.68 855 136664
newbler.refMapper.K12.redo 534 36 41 51 85 1811 116.04 245 61965
454 paired (12.86X cvg) on SFF files
# contig stats
elem min q1 q2 q3 max mean n50 sum q.0cvg r.0cvg 1.0cvg 1.snps 1.breaks.all 1.breaks.1k+
CA.6.1 351 6 3713 8594 17364 83842 13135 23470 4610499 47286 28839 54065 658 31 2
newbler.2.3 477 1069 3507 6708 13167 61181 9287 14368 4429856 212847 160916 212847 268 13 3
newbler.2.5 549 526 2619 5701 11142 72211 8176 13560 4488866 157609 116612 157609 226 11 1
# scaff stats
elem min q1 q2 q3 max mean n50 sum q.0cvg r.0cvg 1.0cvg 1.snps 1.breaks.all 1.breaks.1k+
CA.6.1 14 30345 67631 110633 659844 1216418 331422 686116 4639911 47272 28832 54051 660 366 10
newbler.2.3 17 2023 5481 126476 204123 1958493 275782 705166 4688295 212847 160916 212847 268 474 101
newbler.2.5 13 2060 36701 144055 517319 2057185 364272 711945 4735538 157609 116612 157609 226 548 61
Illumina.36len.52X
- Input reads
6,725,378 x 36bp
- DeNovo assembly
# ctg stats
ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks
edena 7165 100 224 417 744 4375 568 819 4073189 6725378 4983572 591013 95 1
velvet(k=21)* 1694 41 78 988 3856 35320** 2692 7060* 4561195 6725378 2662898 10859 973 8
velvet1.cor(k=21)** 1499 41 71 905 4390 34836* 3043 8038** 4562221 6725378 # Oct2010 :longer
SOAPdenovo 5743 24 27 129 1046 13161 812 2503 4668239 6725378 3688149 99800 692 1
SOAPdenovo.cor 3755 24 27 69 1390 30790 1232 4941 4628933 6725378
- Reference based assembly(Ecoli K12)
ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks AMOScmp 14 674 68206 245583 547319 1199735 331493 548739 4640911 6725378 4099119 16 1417 4 # only 3664073(54%) of seqs aligned to ref => 28X cvg maq** 6 765 145073 727066 1444956 1976524 773274 1444956 4639645 6725378 4116513 23 23 0
- Reference based assembly(Ecoli ATCC) : sharp decrease in assembly quality
ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks AMOScmp* 1530 36 95 532 2933 50248 2828 10506 4328273 6725378 4362159 329400 2837 29 # only 3281823(48%) of seqs aligned to ref => 25X cvg maq 1600 14 100 512 2670 51703 2694 10283 4311361 6725378 4415635 349521 1047 37
- Merged assembly
. elem min q1 q2 q3 max mean n50 sum velvet 1694 41 78 988 3856 35320 2692 7060 4561195 AMOScmp.ATCC 1530 36 95 532 2933 50248 2828 10506 4328273 velvet-AMOScmp.ATCC 418 41 100 1110 9918 208338 11292 51437 4720083
- Repeat stats
elem min q1 q2 q3 max mean n50 sum genome 701 36 42 57 132 3903 197.46 848 138420 edena 57 36 53 75 156 756 139.07 244 7927 SOAPdenovo 130 36 48 67 139 570 118.14 202 15358 velvet* 182 36 48 65 139 855 120.25 229 21885 AMOScmp.K12 681 36 42 59 132 3903 200.68 855 136664 maq.K12 670 36 42 60 140 3903 203.30 855 136213 AMOScmp.ATCC 590 36 42 57 112 3026 164.80 501 97230 ??? max repeat not assembled maq.ATCC 579 36 42 58 122 2029 164.34 479 95154
- Comments
- ACGATGTGACGTACGCGTATGCTCGTATACACACGC appears more than 231K times in the input; does not align to Ecoli or any other genomes
Illuminap.37len.178ins.79X
- DeNovo assembly
# ctg stats
ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks
SOAPdenovo 33346 24 24 25 124 6125 155.08 596 5171181 9619686 7111896 84198 1929 3
velvet 8080 45 178 364 721 7544 561.01 908 4532926 9619686 6199264 95868 1214 2
Illuminap.101len.150ins.173X
- INPUT:
reads: 8,000,000 mates: 4,000,000 clr: 101bp
- OUTPUT:
#ctg stats: . elem min q1 q2 q3 max mean n50 sum CA(OBT) 747 207 2204 4114 7508 35940 5962.97 8812 4454336 SOAPdenovo 2789825 100 101 101 101 1116 101.32 101 282665113 velvet 73160 45 45 45 88 2282 87.06 117 6369253
#scf stats
elem min q1 q2 q3 max mean n50 sum
CA 601 1016 2533 4817 9024 38235 7416.40 11317 4457256
SOAPdenovo 2789825 100 101 101 101 1116 101.32 101 282665113
velvet
Illuminap.101len.150ins.59X.trimmed
- Quality trimming:
- N trimming: reads which contain N's were discarded
- Q20 trimming: beginning/end bases Q20- were deleted
- INPUT:
reads: 3,013,500 mates: 1,506,750 avgClr: 91bp #ctg stats: . elem min q1 q2 q3 max mean n50 sum CA 916 211 1989 3357 5990 44653 4712.96 6317 4317070 SOAPdenovo 64582 100 100 101 104 2209 147.10 101 9500101 velvet(k31) 915 61 238 2191 7008 48470** 4970.98 12083** 4548449
#scf stats
elem min q1 q2 q3 max mean n50 sum
CA 738 1023 2278 4135 7301 44653 5854.51 8275 4320630
SOAPdenovo 60942 100 100 101 101 5705 168.40 125 10262880
velvet(k31) 151 633 6518 10088 18197 48338** 13072.10 18197** 1973887
Illuminap.101len.150ins.150X.cor
- INPUT
reads: 8,835,962 # 20,635,060 reads originally mates: 4,417,981 avgClr: 79bp mea: 150
#ctg stats . elem min q1 q2 q3 max mean n50 sum 0cvgSum snps breaks rearrangements CA 470 573 2986 6181 12285 68568 9614.45 15688 4518792 100041 348 20 3 SOAPdenovo100+ 596 100 260 3233 10238 73870 7609.43 18141 4535220 91689 25 1 velvet100+ 262 103 374 5487 26564 221091** 17346.07 46409** 4544670 14018 405 5
# scf stats . elem min q1 q2 q3 max mean n50 sum rearrangements CA 378 1017 3334 7108 15206 70497 11959.34 21457 4520632 3 SOAPdenovo 165 100 252 2713 35162 410074 27923.05 94982 4607303 0 velvet100+ 174 103 198 1919 30793 413929** 26175.74 95373** 4554578 4
- Reference based assembly(Ecoli K12)
ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks AMOSCmp.soap 8 769 299915 345273 1100345 1444961 579962 1100345 4639697 # soap2 -r 2 -v 2 -n 0 -l 28
- Reference based assembly(Ecoli ATCC)
ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks AMOSCmp.soap100+ 1024 100 431 1348 4811 56348 4199 11899 4300027 # soap2 -r 2 -v 2 -n 0 -l 28 => 0cvg: 1033 regions; 419859 bp AMOSCmp.nucmer100+ 428 100 210 1246 12466 233619 10142 35998 4341162 # nucmer -c 40 -l 20 -b 200 -g 90 => 0cvg: 241 regions; 408109 bp
- Merged assembly
. elem min q1 q2 q3 max mean n50 sum velvet100+ 262 103 374 5487 26564 221091** 17346 46409** 4544670 AMOSCmp-ATCC.nucmer100+ 428 100 210 1246 12466 233619 10142 35998 4341162 velvet-AMOScmp.ATCC.nucm100+ 104 114 1382 14570 52256 413910 45545 121678 4736715
IlluminaTechnicalNotes (Illumina results)
- Metrics: N50 , Max, Genome coverage
- Factors
- Cvg: no improvement beyond 50X :
- ReadLen: reads beyond 36bp(100bp exp) don't help
- Inserts: insert should be longer than longest repeat
insertLen readLen readCvg elem maxScf* n50Scf** genomeCvg*** #readsSampled #mateSampled 200 2*75 50X ? 223,000 97,000 99.58 3,045,862(49X) 1,522,931 200+6K 2*75+2*35 50X+28X ? 2,100,000 1,300,000 99.07 4,099,798(31X) 2,049,899 200+9K 2*75+2*35 50X+28X ? 4,500,000 4,500,000 99.69 3,736,240(28X) 1,868,120
- Quality: best results for N/Failed_Chastity/s35 filter
IlluminaTechnicalNotes (CBCB results)
- Compare given qualities with the computed ones
- sample reads
~/bin/sampleFastq12.pl -r 0.0070 -noN -count 10000 ../s_6_1_export.sample.fastq ../s_6_2_export.sample.fastq Ecoli_1.fastq Ecoli_2.fastq fastq2seq.pl < Ecoli_1.fastq >! Ecoli_1.seq
cat original.count #fwd rev mates s_2_1_3kb_sequence.txt s_2_2_3kb_sequence.txt 21563283 s_2_1_8kb_sequence.txt s_2_2_8kb_sequence.txt 198377 ... cat original.count | p 'next if(/^#/); $c=10000; $r=(($c*1.05)/$F[2]); print "~/bin/sampleFastq12.pl -r $r -count $c original/$F[0] original/$F[1] original_sample/$F[0] original_sample/$F[1]\n";'
- given qualities => avg error
cat Ecoli_1.fastq | fastx_quality_stats | ~/bin/quality2error.pl -i 5 | nl >! Ecoli_1.fastq.pos_qual cat Ecoli_1.fastq.pos_qual | getSummary.pl -i 1 | perl ~/bin/transpose.pl | grep mean => 0.0019493
- estimate qualities : based on alignment to reference : ~ 0.2% (probably lower than it is)
- alignments snps => avg error
nucmer -maxmatch -l 12 -c 12 ~/Escherichia_coli/Data/1con/Ecoli.K12.1con Ecoli_1.seq -p Ecoli_1 cat Ecoli_1.delta | delta-filter-max.pl | ~/bin/deltaextend.pl | show-snps.pl -1 -H | count.pl -p 10000 -i 3 | sort -n >! Ecoli_1.align.pos_qual perl -e '$n =`wc -l Ecoli_1.filter-q.extended.snps | cut -f1 -d " "`; $e=($n/(75*10000)); print $e,"\n" => 0.022



- Estimate library insert sizes
total mated single single.both.aligned single.both.compressed single.both.expand 210 20000 17780 783 30 12 18 6k 20000 14360 3222 1434 560 856 9k 20000 14130 3508 1906 744 1156
- Error correction:
quake.py -f Ecoli.ls -k 18 -p 16
cat Ecoli.ls
Ecoli_1.fastq Ecoli_2.fastq
Ecoli_1.6k.fastq Ecoli_2.6k.fastq
Ecoli_1.9k.fastq Ecoli_2.9k.fastq
original(mates) corrected(mates) corrected(fwd) corrected(rev) deleted
1,522,931 1,241,547 138,323 88,984 335,461
2,049,899 1,665,847 196,868 171,832 399,404
1,868,120 1,528,727 173,005 146,659 359,122
- velvet
#scf100+ stats . elem min q1 q2 q3 maxScf* mean n50Scf** sum genomeCvg*** 200 177 100 168 1427 31804 325,889 25729 95,446 4554080 99.91 200+6K 79 100 121 182 623 1,591,085 59027 1,590,440 4663117 200+9k 78 100 130 190 734 1,637,906 59820 1,093,506 4665962
#ctg100+ stats insertLen elem min q1 q2 q3 maxCtg mean n50Ctg sum 200 312 100 596 5818 22311 110,215 14579 36,329 4548539 200+6K 259 100 475 7990 28357 165,821 17911 42,905 4638953 200+9k 248 100 537 8388 25761 186,539 18648 45,601 4624717
# cvg computed using : nucmer -maxmatch Ecoli.1con Ecoli.ctg.fasta -p 1con-ctg ; delta2cvg.pl < 1con-ctg.delta -M 0 | getSummary.pl -i 4
- CA
#scf100+ stats . elem min q1 q2 q3 maxScf mean n50 sum 200 389 1037 3683 7477 13796 71,906 10484 15837 4078105 # minRead=64;minOvl=40;minUtg=1000 (default) 200.redo 420 208 2443 4372 8100 39,715 6343 9202 2664254 # minRead=32;minOvl=30;minUtg=500 200+6k ? # minRead=32;minOvl=30;minUtg=500 : run for a very long time ; killed 200+6k.redo2 166 75 1298 1904 8579 101,429 6257 14354 1038602 # minRead=32;minOvl=30;minUtg=500;utgErrorRate=0.03 ; lots of degenerates
#ctg100+ stats . elem min q1 q2 q3 maxCtg mean n50 sum 200 657 104 2570 4773 8167 39,986 6199 8626 4072659 200.redo 454 75 2322 4153 7505 35,904 5866 8744 2663112 200+6k ? # minRead=32;minOvl=30;minUtg=500 : run for a very long time ; killed 200+6k.redo2 612 35 215 1132 1827 4,994 1195 1904 731055 # minRead=32;minOvl=30;minUtg=500;utgErrorRate=0.03 ; lots of degenerates
- Note
- Shorter velvet ctgs/scf on corrected reads: the input reads are already high quality !!!
IlluminaTechnicalNotes sim data err=0.0
insertLen readLen readCvg 200+6K 2*75+2*35 50X+28X 200+6K 2*75+2*75 50X+28X
- velvet
#scf100+ stats . elem min q1 q2 q3 max mean n50 sum velvet.35 72 100 133 182 531 4636779 64823 4636779 4667231 velvet.75 72 100 129 182 531 4636870 64822 4636870 4667211
#ctg100+ stats . elem min q1 q2 q3 max mean n50 sum velvet.35 154 100 191 1919 42330 263156 30158 93526 4644318 velvet.75 145 100 178 1331 45218 263151 32092 98728 4653398
- CA
- CA.35 : cgw would take days to complete; Solution: delete 526,644 reads that assemble into single fragment unitigs (7% of total reads)
#scf100+ stats . elem min q1 q2 q3 max mean n50 sum CA.35 20 7383 81163 122901 359340 729797 222118 409560 4442362 CA.75 1 4640849 4640849
#ctg100+ stats CA.35 97 6687 14090 29769 57870 326996 44421 67366 4308861 CA.75 47 102 14731 68369 128240 515019 98614 176651 4634873
Illuminap.64len.475ins.50X
- Simulated data : maq simulator :
maq simulate -d 475 -s 47 -1 64 -2 64 -N 1812500 -r 0.001 Ecoli_1.fastq Ecoli_2.fastq Ecoli.1con Ecoli.simupars.dat soap2 -D Ecoli.1con.index -a Ecoli_1.fastq -b Ecoli_2.fastq -M 0 -o Ecoli_12.mated.soap2 -2 Ecoli_12.single.soap2 -m 375 -x 575 -p 20 -r 2 cat *soap2 | ~/bin/AMOS/soap2posmap.pl | ~/bin/posmap2cvg.pl -M 0 | getSummary.pl -i 4 . elem min q1 q2 q3 max mean n50 sum 0cvg 1382 1 2 3 5 84 4.71 5 6512 #due to mutation
reads: 3,625,000 clr: 64 bp mea;std : 475;47
#ctg stats . elem min q1 q2 q3 max mean n50 sum velvet100+ 261 100 441 7429 25373 152351 17468 41524 4559162 CA.ctg100+ 0 #no ctg/scf just deg max 200; is cvg too low? #scf stats velvet100+ 148 100 158 923 33253 327108 30850 132780 4565870
- Reference based assembly(Ecoli K12)
elem min q1 q2 q3 max mean n50 sum AMOSCmp.nucmer100+ 1 4639681 4639681
- Reference based assembly(Ecoli ATCC)
elem min q1 q2 q3 max mean n50 sum AMOSCmp.nucmer100+ 510 100 296 1741 11087 105786 8495 27761 4332605 # nucmer -c 28 -l 14 => 0cvg: 334 regions; 401485 bp
- Merged assembly
elem min q1 q2 q3 max mean n50 sum velvet-AMOSCmp.nucmer100+ 138 114 1861 17205 50382 268321 39660 105786 5473097 # total length is >> genomeLength (some duplication going on)
Illuminap.64len.475ins.50X-64len.8000ins.10X (60X cvg)
reads: 3,625,000 + 725,000 clr: 64 bp mea;std : 475;47 8000;800
#ctg stats . elem min q1 q2 q3 max mean n50 sum SOAPdenovo100+ 576 100 249 3249 9909 73,093 7893 21083 4546797 velvet100+ 179 100 223 2955 35078 240,045 25933 83796 4642027
#scf stats SOAPdenovo100+ 241 100 151 730 6746 492338 19669 219830 4740402 # pair_num_cutoff=default SOAPdenovo100+ 213 100 114 151 296 2,142,392 22266 1430222 4742568 # pair_num_cutoff=50 velvet100+ 75 100 133 204 623 4,406,037 62160 4406037 4661969
Staphylococcus aureus
- SRA Study Staphylococcus aureus Sequencing on Illumina
- 22 complete strains: NCBI search: "Staphylococcus aureus"[Organism] AND pt_default[prop] AND seqstat_complete[PROP]
- /fs/szdata//ncbi/genomes/Bacteria/Staphylococcus_aureus_*/
- chromosome lengths 2.7-2.9Mbp
- plasmids? (2-3)
- Ref:
id len gc% desc NC_010079 2872915 32.76 Staphylococcus aureus subsp. aureus USA300_TCH1516, complete genome (most related to Illumina data set) NC_003923 2820462 32.83 Staphylococcus aureus subsp. aureus MW2, complete genome NC_007622 2742531 32.78 Staphylococcus aureus RF122, complete genome NZ_AASB00000000 2810505 32 Staphylococcus aureus subsp. aureus USA300_TCH959, 256 contigs
USA300-MW2 . elem min q1 q2 q3 max mean n50 sum %id 2873390 79.28 98.93 99.30 99.54 100.00 98 99 284357849.9 0cvg 124 2 60 430 1357 18282 1149 2539 142568
- Repeats (Saureus USA300)
. elem min q1 q2 q3 max mean n50 sum 36bp+ 667 36 44 62 98 5016 125 191 83788 100bp+ 158 100 119 158 254 5016 348 746 55049 200bp+ 56 201 253 324 746 5016 735 1506 41185
- Uniq (Saureus USA300)
. elem min q1 q2 q3 max mean n50 sum 36bp+ 623 1 10 75 820 126818 4467 34787 2783139 100bp+ 142 1 76 1512 32659 180152 19801 74137 2811878 200bp+ 48 1 221 35708 88217 328320 58812 145211 2823011
Read stats
. reads min max mean n50 sum cvg SRA accession(s) 454 391,495 51 857 273 275 107151210 37.29 SRX002328 strain USA300_TCH959 HMP0023 454 paired 490,520 1 1323 134 182 65822848 22.95 SRX002327 strain USA300_TCH959 HMP0023 ; NOMINAL_LENGTH=2750 NOMINAL_STDEV=250 Illumina 2,739,566 36 36 36 36 98624376 34.32 SRX007668 qual plot Illumina paired end 8,982,084 36 36 36 36 323355024 112.55 SRX007696 qual plot # NOMINAL_LENGTH=175 Illuminia jumping lib 17,678,908 37 37 37 37 654119596 227 SRX007711 # NOMINAL_LENGTH=300, ORIENTATION=5'3'-3'5'; soap estimate 3538 Illumina paired 30,597,352 101 101 101 101 3090332552 1075 SRX007714 # NOMINAL_LENGTH=300, NOMINAL_SDEV=22.253, ORIENTATION=5'3'-3'5 ; CA estimate mea/std=170/21; strain USA300_TCH1516
{kind=link}
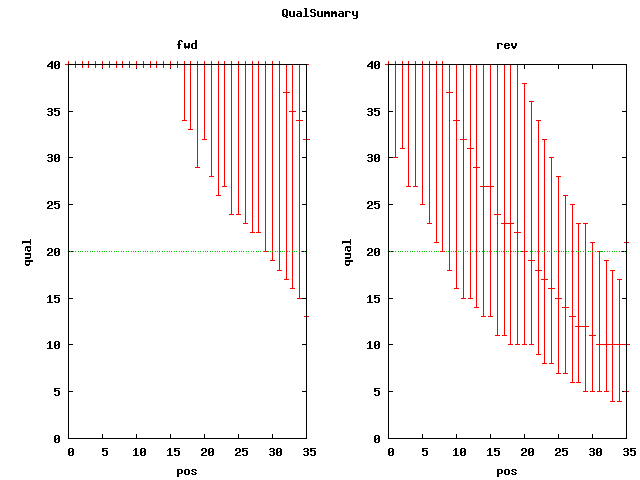{kind=link}
454 stats (37.29X read cvg)
- DeNovo
. ctgs min q1 q2 q3 max mean n50 sum CA.6.0.bog 31 1471 16316 51985 142506 318772 89171 157559 2764330 CA.6.1.bog 47 1066 6623 26392 73408 309139 58908 145306 2768666 # fewer degenerates compared to CA.6.0.bog
newbler.2.3.deNovo 85 104 203 1176 19635 295958 32888 157201 2795541 newbler.2.5p1.deNovo* 46 105 231 3087 56818 546353* 60730 296002* 2793599
- Reference based(Saureus USA300_TCH1516 (diff strain))
. ctgs min q1 q2 q3 max mean n50 sum newbler.2.3.refMapper 183 103 630 3737 18738 117468 14374 42301 2630491 newbler.2.5p1.refMapper 186 103 589 3740 17184 117464 14139 43130 2629763
454 paired stats (22.95X read cvg)
- Data
reads min q1 q2 q3 max mean n50 sum sff(original) 334241 36 201 256 277 362 235 264 78432227 sffToCA(adaptor free) 325555 51 103 148 208 358 157 185 51225615 # 58723 mates
- DeNovo
# ctg stats . ctgs min q1 q2 q3 max mean n50 sum CA.6.0.bog 11 278 47961 194288 508135 590074* 255272 508135* 2808001 CA.6.1.bog 18 238 21014 135971 239466 567548 155836 277888 2805055 newbler.2.3.deNovo 544 100 810 2652 7672 33434 5126 10865 2788673 newbler.2.5p1.deNovo 100 103 295 3287 39467 229053 27879 78379 2787870
# scf stats . scfs min q1 q2 q3 max mean n50 sum CA.6.0.bog 6 278 47961 194288 1035529 1410221 468016 1410221 2808101 CA.6.1.bog 6 284 21014 173065 1032129 1458733 467554 1458733 2805325 newbler.2.3 16 2042 2173 2767 110212 1409442 176373 1037981 2821966 newbler.2.5p1.deNovo 8 2475 20731 110137 1030785 1408642 349895 1408642 2799157
- Reference based(Saureus USA300)
. ctgs min q1 q2 q3 max mean n50 sum newbler.2.3.refMapper 206 103 556 3098 15366 117487 12749 40687 2626469
Illumina stats (34.32X read cvg)
- DeNovo
. ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks edena 3956 100 207 422 851 13908 656 1040 2597617 2739566 ? 315399 18 1 SOAPdenovo* 5814 24 45 164 624 13087 503 1331 2925517 2739566 ? 69547 298 1 velvet 3477 45 166 464 1054 10468 812 1528 2825440 2739566 ? 100722 638 17
- Reference based(Saureus USA300)
. ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks AMOScmp 81 36 5859 20119 52879 196907 35474 75670 2873401 2739566 1003517 376 649 3 # only 2126008(77%) of seqs aligned to ref => 26X cvg maq** 41 36 10188 33799 114655 341537 70064 154157 2872626 2739566 955336 369 43 0
- Reference based(Saureus MW2)
. ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks AMOScmp 1199 36 88 366 2000 48748 2268 8866 2720299 2739566 1118637 152285 2369 19 # only 1965762(71%) of seqs aligned to ref => 24X cvg maq* 1052 36 91 364 2111 64379 2576 10270 2710270 2739566 1100695 167184 2357 21
Illumina paired stats (112.55X read cvg; avgInsert=~60bp)
- Read alignments
73% of the reads align by "soap -g 5 -v 5 -r 2 -f 0" 3368102 fwd 3210408 rev 6578510 total
- DeNovo
#ctg stats . ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks edena 4636 100 216 388 707 6754 544 782 2526187 2739566 ? 385985 88 2 # most 0cvg regions SOAPdenovo 6038 24 26 34 47 46791 498 11310 3009291 8982084 ? 48023 38 0 velvet** 705 45 60 84 584 142306 4046 33811 2852996 8982084 2898483 39096 854 12
#scf stats . scfs min q1 q2 q3 max mean n50 sum SOAPdenovo 482 100 270 1532 6719 58192 5849 18859 2819512 velvet 12 333 1111 27392 53828 142405 31488 78092 377866 # 2+ctg scaffolds
- Reference based(Saureus USA300)
. ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks maq** 1 2872913 2872913 2872913 2872913 2872913 2872913 2872913 2872913 8982084 2614519 2 3 0 Comments: * assembles in one piece * first & last bases in ref not covered * there is no read = ref.1-36 or ref.2872880-2872915
- Reference based(Saureus MW2)
. ctgs min q1 q2 q3 max mean n50 sum seqs singl 0cvgSum snps breaks maq 695 36 89 399 3304 77110 3914 17814 2720683 8982084 2801829 158246 4132 57
Illumina101 paired stats (cvg 78X; givenInsert=300 ; computerInsert=170)
- DeNovo
# ctg stats . ctgs min q1 q2 q3 max mean n50 sum CA.bog 73 1716 14503 26431 52086 148524* 39043 58038* 2850157 SOAPdenovo 9382 32 32 52 63 85850 347 16726 3259522 velvet 453 61 85 224 3279 137163 6297 36496 2852552
# scf stats . scf min q1 q2 q3 max mean n50 sum CA.bog 67 1716 14869 32929 58038 148524* 42541 65383* 2850277 SOAPdenovo 186 100 333 1528 17623 144079 15625 55558 2906207 velvet 427 61 82 177 2982 137163 6685 37874 2854649
- Comparative
Pseudomonas aeruginosa
- PLoS article
From 8,627,900 reads, each 33 nucleotides in length, we assembled the genome into one scaffold of 76 ordered contiguous sequences containing 6,290,005 nucleotides, including one contig spanning 512,638 nucleotides, plus an additional 436 unordered contigs containing 416,897 nucleotides.
Read stats
. elem min q1 q2 q3 max mean n50 sum cvg original 8627900 33 33 33 33 33 33.00 33 284720700 43.55X cvg cor(quake) 7401074 30 33 33 33 33 32.75 33 242393133 37X cvg
Assembly stats
- DeNovo
elem min q1 q2 q3 max mean n50 sum velvet(k=23) 7821 45 214 502 1108 16239 862.12 1541 6742621 !!! previous velvet1.cor(k=21)** 5946 41 179 574 1472 20233 1129.77 2362 6717612 !!! new best
- Comparative PA14
. elem min q1 q2 q3 max mean n50 sum AMOScmp.nucmer 1843 20 41 63 325 127984 3350.39 30660 6174765 !!! previous best : nucmer aligner AMOScmp.soap 1929 31 59 223 3395 79432 3182.93 13233 6139867 !!! new : soap2 aligner AMOScmp.bowtie 1701 31 56 215 3535 79443 3610.23 15916 6141006 !!! new : bowtie aligner
- Merged
velvet1.cor(k=21)** 5946 41 179 574 1472 20233 1129.77 2362 6717612 !!! new best AMOScmp.nucmer 1843 20 41 63 325 127984 3350.39 30660 6174765 !!! previous best : nucmer aligner AMOScmp.nucmer-velvet 983 41 174 796 2859 309985 7390.19 53631 7264555
SRA projects
Whole genome based sequencing of a Escherichia fergusonii strain using 454 Technology.
- SRP001132
- 1 complete strain
- Use 454 single/paired , titanium
E coli Whole Genome Sequencing on 454 and Illumina
- Mentioned by allpaths-lg
- SRP001087
Genome Sequencing of Mycobacterium tuberculosis PGG1
Plasmodium falciparum Whole Genome Assembly Development
- Mentioned by allpaths-lg
- SRP001775
- ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/litesra/SRP/SRP001/SRP001775/
Rhodobacter sphaeroides Sequencing on Illumina
- Mentioned by allpaths-lg
- SRP001079
- 4 complete strains: NCBI search: "Rhodobacter sphaeroides"[Organism] AND pt_default[prop] AND seqstat_complete[PROP]
- /fs/szdata//ncbi/genomes/Bacteria/Rhodobacter_sphaeroides_*/
- chromosome lengths ~ 4Mbp
- several plasmids
- largest repeat : 5348 at 99.93%id; other repeats < 3k
Staphylococcus aureus Sequencing on Illumina
Whole Genome Sequencing of Streptomyces roseosporus NRRL 11379
Streptococcus pyogenes pathogenicity
- SRP000775 Broad 38.5% gc
Vibrio Choerae : PacBio long reads !!! =
Simulaterd reads
- use MetaSim
- MetaSim limitations:
- Illumina reads (Empirical error model) always length==36
- Exact reads : always unmated
- get quality values & mate pairs:
~/bin/fasta2qual.pl prefix.seq > prefix.qual
cat prefix.seq | grep "^>" | awk '{print $1}' | grep 1$ | p '/>(.+).1/; print "$1.1 $1.2\n";' > prefix.mates
convert-fasta-to-v2.pl [ -454 ] -l prefix -mean 5000 -stddev 500 -s prefix.seq -q prefix.qual -m prefix.mates > prefix.frg
SRA
Download
- SRRs (runs)
cat SRR.txt | perl -ane 'next unless /(SRR...)/; print "wget ftp://ftp.ncbi.nih.gov/sra/sra-instant/reads/ByRun/litesra/SRR/$1/$F[0]/$F[0].lite.sra\n";' => SRR001355.lite.sra cat SRR.txt | perl -ane 'next unless /(SRR...)/; print "wget ftp://ftp.ncbi.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/$1/$F[0]/$F[0].sra\n";' => SRR001355.sra
- SRXs (experiments)
cat SRX.txt | perl -ane '/next unless (SRX...)/; system "wget -r ftp://ftp.ncbi.nih.gov/sra/sra-instant/reads/ByExp/litesra/SRX/$1/$F[0]/\n";'
- SRPs (studies)
cat SRP.txt | perl -ane '/next unless (SRP...)/; system "wget -r ftp://ftp.ncbi.nih.gov/sra/sra-instant/reads/ByStudy/litesra/SRX/$1/$F[0]/\n";'
Convert
cat SRR.txt | perl -ane 'print "fastq-dump -A $F[0] -D $F[0].*sra\n";' => SRR??????_[12].fastq cat SRR.txt | perl -ane 'print "sff-dump -A $F[0] -D $F[0].*sra\n";' => SRR??????.sff
- sff-dump dump does not work on litesra
Download and convert
- Example:
~/bin/SRRget.pl [-lite] SRR075011 | sh # => SRR075011.fastq
Insert Size Estimate
soap2-index ../1con/genome.1con touch ../1con/genome.1con.index
cat SRR.txt | perl -ane 'print "soap2 -D ../1con/*.index -a $F[0]_1.fastq -b $F[0]_2.fastq -o $F[0]_12.mated.soap2 -2 $F[0]_12.single.soap2 -p 20\n";' cat SRR.txt | perl -ane 'print "cat $F[0]_12.*.soap2 | ~/bin/AMOS/soap2sameRef.pl -dist -max 10000 | getSummary.pl -i 1\n";' # => q2 ; add q2 vals as 2nd col in SRR.txt
cat SRR.txt | perl -ane '$m=$F[1]*0.7; $x=$F[1]*1.3; print "soap2 -D ../1con/*.index -a $F[0]_1.fastq -b $F[0]_2.fastq -o $F[0]_12.mated.soap2 -2 $F[0]_12.single.soap2 -p 20 -m $m -x $x " ; print "-R" if($F[1]>1000); print "\n";'
454
Format
- sff
- linkers
L454.flx 44 45.45 (palindrome) L454.titanium.fwd 42 30.95 L454.titanium.rev 42 30.95
- fasta
- fastq
Data sets
- Sff
/nfshomes/dpuiu/methanobrevibacter_smithii/Data/454/*sff # single ; FLX ? /nfshomes/dpuiu/F_tularensis/SRA_FTP/*/*sff # single & paired ; GS20 & FLX /nfshomes/dpuiu/Brugia_malayi.new/Data/brugia-sequencing/*/*sff # paired ; FLX & Ti /fs/szattic-asmg4/dpuiu/HTS/Escherichia_coli/Data/454/*fastq # single from SRA !!! /fs/szattic-asmg4/dpuiu/HTS/Escherichia_coli/Data/454p/*fastq # paired from SRA !!!
Converters
- sff -> sff
sffinfo -a in.sff | head -100 > out.acc sfffile -o out.sff -i out.acc in.sff
- sff -> seq/qual (454 package)
sffinfo -s prefix.sff > prefix.seq sffinfo -q prefix.sff > prefix.qual
- sff -> frg2 (wgs package)
sffToCA -insertsize mean std -linker [flx|titanium] -trim chop -libraryname prefix -output prefix.frg2 prefix.sff # => prefix.frg2 -trim chop: the read is chopped down to just the clear bases !!! should try it
- fastq -> seq/qual -> frg2 (dpuiu & wgs)
fastq2seq.pl < prefix.fastq > prefix.seq fastq2qual.pl < prefix.fastq > prefix.qual convert-fasta-to-v2.pl -454 -l prefix -s prefix.seq -q prefix.qual > prefix.frg2
- fastq -> frg2 (dpuiu & wgs)
fastq2frg.sh prefix # => prefix.frg2
- seq/qual -> clean seq/qual -> frg2 (linker removal for 454p) (dpuiu)
454p2seqqual.amos prefix # => prefix.cseq, prefix.cqual convert-fasta-to-v2.pl -454 -l prefix -s prefix.cseq -q prefix.cqual -l prefix -mean 3000 -stddev 300 -m prefix.mates > prefix.frg
See also
Illumina
Formats
- Raw : the intensity values (int) and noise (nse) values.
- Processed : the processed intensity values (sig2) and four-channel quality values (prb).
- Base : the base calls (the quality value is gotten from the prb for the called base).
Converters
- illumina <-> srf (illumina package)
illumina2srf -o lane_no.srf BustardDir/s_lane_no_*_qseq.txt srf2illumina file.srf
- split joined paired reads from SRA
# seq/qual cat prefix.seq | perl ~/bin/splitSeqMiddle.pl > prefix.cseq cat prefix.qual | perl ~/bin/splitSeqMiddle.pl -q > prefix.cqual # fastq cat prefix.fastq | perl ~/bin/splitFastqMiddle.pl > prefix.cfastq cat prefix.fastq | perl ~/bin/splitFastqMiddle.pl -f > prefix.fwd.cfastq cat prefix.fastq | perl ~/bin/splitFastqMiddle.pl -r > prefix.rev.cfastq
SOLiD
Converters
- SOLiD -> srf (SOLiD package)
/fs/szdevel/dpuiu/solid2srf_v0.6.6/
Converters
SFR
Commands:
srf2fasta srf2fastq srf_dump_all srf_extract_hash srf_extract_linear srf_filter srf_index_hash srf_info
FASTA/QUAL
- seq/qual => frg2
convert-fasta-to-v2.pl -l prefix -s prefix.seq -q prefix.qual > prefix.frg2
- seq/qual => amos
toAmos -m prefix.mates -s prefix.seq -q prefix.qual -o prefix.afg
FASTQ
- http://en.wikipedia.org/wiki/FASTQ_format
- The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants
format offset qualityRanges
0 1 2 3 4
01234567890123456789012345678901234567890
===================================================================
Sanger(cont. numbers) 33 !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHI !\"\#\$%&\'\(\)\*\+,-.\/0123456789:;<=>?\@ABCDEFGHI
Illumina(cont. low cases) 64 @ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefgh
CA frg packed 48 0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWX
===================================================================
- fastqToCA
fastqToCA -insertsize 3000 300 -libraryname short -type sanger -fastq $PWD/short_1.fastq,$PWD/short_2.fastq > short_12.frg runCA -d . -p genome stopAfter=initialStoreBuilding short.frg gatekeeper -dumpfrg -allreads -donotfixmates -format2 genome.gkpStore > genome.frg
- fastq : input reads; id positions don't necessarily match; fwd read names end in 1, rev read names end in 2
=> fastq (_0: unmated; _[12]: mated)
fastq2seqs.pl < input_1.fastq | sed 's/1$//' > prefix_1.mate fastq2seqs.pl < input_2.fastq | sed 's/2$//' > prefix_2.mate intersect.pl prefix_1.mate prefix_2.mate > prefix.mate cat prefix.mate | perl -ane 'print "$F[0]1 $F[0]2\n";' > prefix.mates difference.pl prefix_1.mate prefix.mate | perl -ane 'print "$F[0]1\n";' > prefix.unmated difference.pl prefix_2.mate prefix.mate | perl -ane 'print "$F[0]2\n";' >> prefix.unmated extractfromfastqnames -i 0 -f prefix.unmated < prefix.fastq > prefix_0.fastq extractfromfastqnames -i 0 -f prefix.mates < prefix.fastq > prefix_1.fastq extractfromfastqnames -i 1 -f prefix.mates < prefix.fastq > prefix_2.fastq
#or fastq22fastq3.amos -D FWDSEQ=input_1.cor.txt -D REVSEQ=input_2.txt prefix
- fastq2fastq
maq sol2sanger illumina.fastq sanger.fastq ~/bin/fastq2afstq -in illumina -out sanger < illumina.fastq > sanger.fastq ~/bin/fastq2afstq -in sanger -out illumina < sanger.fastq > illumina.fastq
- fastq => frg2
fastq2seq.pl < prefix.fastq > prefix.seq fastq2qual.pl < prefix.fastq > prefix.qual convert-fasta-to-v2.pl -l prefix -s prefix.seq -q prefix.qual > prefix.frg2 ~/bin/fastq2frg.sh prefix
FRG
- frg => frg2
convert-v1-to-v2.pl [-v vector-clear-file] [-noobt] [-unmated] < prefix.frg > prefix.frg2
- frg => amos
toAmos -f prefix.frg -o prefix.afg
- frg2 => frg
gatekeeper -o prefix.gkpStore -T prefix.frg2 gatekeeper -dumpfrg prefix.gkpStore > prefix.frg
- frg => mates
frg2mates.pl < prefix.frg > prefix.mates frg2acc.pl < prefix.frg > prefix.acc cat prefix.acc | ~/bin/difference1or2.pl -j1 1 -f prefix.mates > prefix.unmated
extractfromfastqnames -i 0 -f prefix.unmated < prefix.fastq > prefix_0.fastq extractfromfastqnames -i 0 -f prefix.mates < prefix.fastq > prefix_1.fastq extractfromfastqnames -i 1 -f prefix.mates < prefix.fastq > prefix_2.fastq
AMOS
- amos => frg
amos2frg -i prefix.afg -o prefix.frg [-a accession] removefromfrgmsg.pl prefix.frg ADT # in case convert-v1-to-v2.pl is run afterwards
- amos => bank
bank-transact -c -z -b prefix.bnk -m prefix.afg
# for velvet assemblies
grep "^>" Sequences | sed 's/>//' | awk '{print $1,$2}' > prefix.seqs
bank2contig prefix.bnk/ | grep ^# | grep -v ^## | p '/(\d+)/; print $1,"\n";' | > prefix.assembled
difference.pl -i 1 prefix.seqs prefix.assembled > prefix.singletons
TA (seq,qual,xml)
- seq,qual,xml => frg
tarchive2ca -o prefix -c prefix.clr -l prefix.libinfo prefix.seq # => prefix.frg tracedb-to-frg.pl (?)
- seq,qual,xml => amos
tarchive2amos -o prefix -c prefix.clr -l prefix.libinfo prefix.seq # => prefix.afg
ACE
- ace => amos
toAmos -ace prefix.ace -m prefix.mates -o prefix.afg
Input processing
Quality assessment
- Daniela's scripts
cat prefix_1.fastq | ~/bin/getQualSummary.pl -type illumina > prefix_1.qual.summary cat prefix_2.fastq | ~/bin/getQualSummary.pl -type illumina > prefix_2.qual.summary cat ~/bin/plotQualSummary.gp | sed 's/prefix/Ecoli/g' >! plotQualSummary.gp gnuplot plotQualSummary.gp display Ecoli.qual.summary.png
- Galaxity FASTX scripts
~/bin/fastx.sh prefix or fastx_quality_stats -i prefix.fastq -o prefix.stats fastq_quality_boxplot_graph.sh -i prefix.stats -o prefix.quality_boxplot.png fastx_nucleotide_distribution_graph.sh -i prefix.stats -o prefix.nucleotide_distribution_graph.png display *.png
#the counts should be pretty uniform ls s*stats | p 'print "getSummary.pl -i 12 -t $F[0] -nh <$F[0]\n";' | sh | pretty > A_count.stats ls s*stats | p 'print "getSummary.pl -i 13 -t $F[0] -nh <$F[0]\n";' | sh | pretty > C_count.stats ls s*stats | p 'print "getSummary.pl -i 14 -t $F[0] -nh <$F[0]\n";' | sh | pretty > G_count.stats ls s*stats | p 'print "getSummary.pl -i 15 -t $F[0] -nh <$F[0]\n";' | sh | pretty > T_count.stats
Coverage assesment
soap2 .... cat prefix*soap2 | ~/bin/AMOS/soap2posmap.pl | ~/bin/posmap2cvg.pl > prefix.cvg plot "prefix.cvg" u 2:6 w l
Read trimming
- Delete all reads that contain an N (or a quality of 0)
- Compute Q20 & trim all reads to that length
- Compute nucleotide bias. Trim 5',3' ends that show bias
- Trim 5',3' ends
Adaptor removal
- fastx_clip -a adaptorSeq !!! only frims the 3'
- https://github.com/chapmanb/bcbb/blob/master/align/adaptor_trim.py
- check high frequency kmers
- sometimes there is adaptor in short insert size libs
- Using regular expressions (tested)
~/bin/cleanTitaniumAdaptorFromFastq.pl ~/bin/grep.pl -f ~/bin/titanium.fgrep ~/bin/cleanAdaptorFastq.pl -tita -min 32
Read correction
- quake
echo prefix_1.fastq prefix_2.fastq > prefix.ls quake.py -f prefix.ls -k 18 -p 16
or
cat prefix*fastq | count-qmers -k 18 > prefix.ls.qcts gzip prefix.ls.qcts cov_model.py prefix.ls.qcts | tail -1 | p 'print $F[1]' > prefix.cutoff # takes pretty long to run cov_model.py 18mers.qcts >& /dev/null tail -1 cutoff.txt | perl -ane 'print $F[0]' > prefix.cutoff correct -f prefix.ls -k 18 -c `cat prefix.cutoff` -m prefix.ls.qcts -p 4
- Aleksey's
set SIZE=100000000 # 100M jellyfish count -s 1000000 -m 18 --both-strands -t 16 -o frag.k18.jf frag_?.*fastq jellyfish merge frag.k18.jf_* -o frag.k18.jf jellyfish histo frag.k18.jf | head -20 => bottom=cuttof correct -f prefix.ls -k 18 -c cutoff -m 18mers.qcts -p 4
- SOAP correction
ls -1 prefix_1.fastq prefix_2.fastq > prefix.ls ~/szdevel/correction/KmerFreq -i prefix.ls -o prefix.cor -s 18 ~/szdevel/correction/Corrector -i prefix.ls -r prefix.cor.freq -s 18 -t 16
Issue: too many reads are discarded
- hybrid-shrec : multiple platform correction
http://www.cs.helsinki.fi/u/lmsalmel/hybrid-shrec/
Lucy
lucy -debug prefix.lucy.info prefix.seq prefix.qual
cat prefix.lucy.info | awk '{print $1,$3,$4}' > prefix.lucy.clr
~/bin/clrFasta.pl -f prefix.lucy.clr < prefix.seq > prefix.lucy.seq
~/bin/clrFasta.pl -f prefix.lucy.clr -q < prefix.qual > prefix.lucy.qual
CA
- 454 reads
runCA -stopAfter OBT ... gatekeeper -dumpfragments -tabular asm.gkpStore | p 'print $F[-3],"\n" unless($F[6]);' | getSummary.pl -t ORIG gatekeeper -dumpfragments -clear CLR -tabular asm.gkpStore | p 'print $F[-1]-$F[-2],"\n" unless($F[6]);' | getSummary.pl -t CLR gatekeeper -dumpfragments -clear OBTINITIAL -tabular asm.gkpStore | p 'print $F[-1]-$F[-2],"\n" unless($F[6]);' | getSummary.pl -t OBTINITIAL gatekeeper -dumpfragments -clear OBTCHIMERA -tabular asm.gkpStore | p 'print $F[-1]-$F[-2],"\n" unless($F[6]);' | getSummary.pl -t OBTCHIMERA gatekeeper -dumpfragments -clear LATEST -tabular asm.gkpStore | p 'print $F[-1]-$F[-2],"\n" unless($F[6]);' | getSummary.pl -t LATEST
Simulators
- Maq: max read len can be recompiled; must be trained on a data set;
~/db/Illumina.simutrain.dat : trained on 75 bp Illumina "high qual" reads (q2>30 for all pos)
maq simutrain train.dat train.fastq maq simulate -d libMea -s libStd -r 0 -N noMates -1 read1Len -2 read2Len read1Out read2Out ref.fasta train.dat
- Samtool
wgsim -e 0 -d libMea -s libStd -r 0 -N noReads -1 read1Len -2 read2Len ref.fasta read1.fastq read2.fastq -C # error free reads wgsim -e readErr -d libMea -s libStd -r 0 -N noReads -1 read1Len -2 read2Len ref.fasta read1.fastq read2.fastq -C # error in reads
- Others:
dwgsim simseq Used by Assemblathon !!!