Francisella tularensis holarctica OSU18
Center: Baylor Status: Complete
Data
Reference:
Genbank Accession: CP000437.1 RefSeq Accession: NC_008369.1 GI: 115313981 Taxname: Francisella tularensis subsp. holarctica OSU18 Taxid: 393011 Genome_ID = 19819 DNA length = 1895727 Genetic Code: 11 Publications: 16980500; Protein count: 1555 CDS count: 1555 RNA count: 49 Gene count: 1932 Others: 329 Total: 3865
NC_008369-NC_008369.png Mummerplot Francisella tularensis holartica OSU18 (complete genome) vs itself : 29,384 bp repeat (99921,129304):(1098847,1128228)
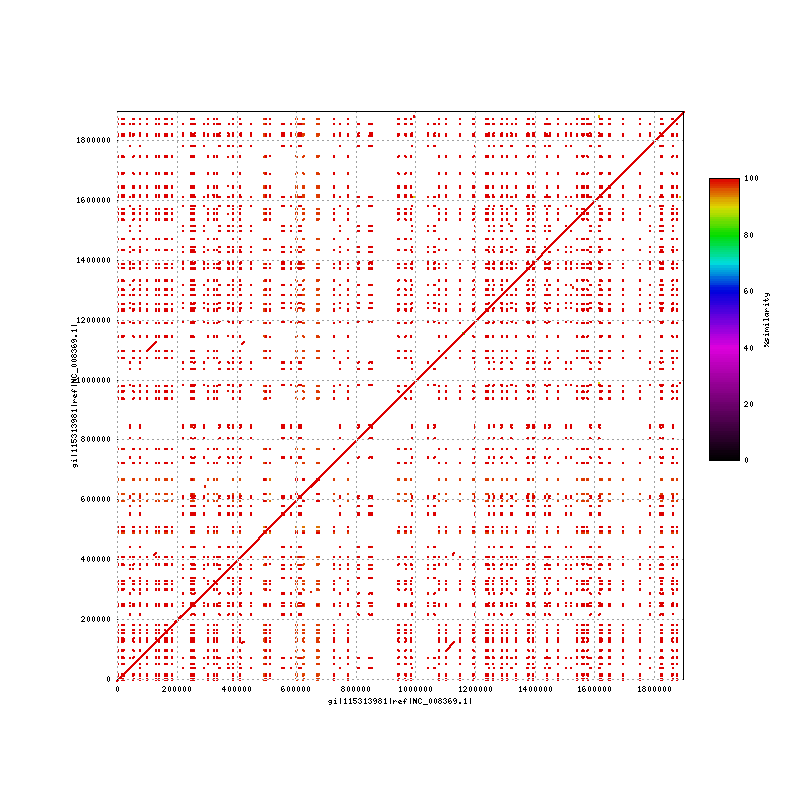{kind=link}
Paper:
Chromosome Rearrangement and Diversification of Francisella tularensis Revealed by the Type B (OSU18) Genome Sequence ; JBacteriol 2006
Genome sequencing and assembly. Sequencing and assembly of the F. tularensis subsp. holarctica strain OSU18 genome were accomplished by the whole-genome shotgun (WGS) method, similar to a previously described method (22). Briefly, the WGS clones were sequenced using ABI 3730 sequencers, and the sequence bases were called using the Applied Biosystems sequencing analysis software KB Basecaller. The WGS reads were assembled by using Atlas (11) and Phrap (7). The initial WGS assembly resulted in 132 contigs in 33 scaffolds with approximately 26× sequence coverage. Gaps between contigs and scaffolds were closed by sequencing PCR products that spanned gaps or by sequencing small insert libraries generated from the PCR products. Low-quality regions were resequenced using clones or PCR products spanning the regions to ensure that the Phrap quality score for each base was equal to or greater than 30. This relatively deep data set should enable further studies involving new sequencing, comparative genomics, and proteomics strategies and technologies. Included among these strategies and technologies are (i) using sequencing reads to scan for possible phase variation in Francisella cultures, (ii) using WGS clones as gene expression constructs for peptide array-based antigen screens, and (iii) using the deep coverage of sequencing reads as a representative data set for comparing existing sequencing methodologies to new technologies in various stages of development.
Traces: from NCBI TA
Libraries:
CENTER PROJECT STRAIN SEQ_LIB_ID TYPE SIZE STDEV COUNT Location Comment BCM BFTB OSU-18 BFTBP WGS 2,000 1,000 58,053 TA acc=3379596; INSERT_SIZE,STD=2690,643 (WGA) BCM BFTD OSU-18 BFTDP WGS 2,000 1,000 10,409 TA acc=3379603; INSERT_SIZE,STD=3675,1407 (WGA) BCM 4WG_FTOS OSU18 4WGS_FTOSA 454 . . 310,747 TA BCM 4WG_FTUL.OS OSU18 4WG_FTUL.OS_000pA 454p . . 216,495 SRA ? StrainSubtotal 595,704 Baylor site: 65,131 Sanger & 317,789 454 Reads
Notes
- 22727 out of 216495 (>10%) 454p don't contain a 100% identical copy of the linker GTTGGAACCGAAAGGGTTTGAATTCAAACCCTTTCGGTTCCAAC
- 687 sequences contained 2 copies of the adapter
Location:
/fs/szasmg/Bacteria/F_tularensis
Assembly
Steps:
1. The complete genome sequence was downloaded from NCBI: NC_008369.1
2. Reads were downloaded from TA and formatted using tarchive2ca
3. Only the 2 Sanger libraries for this project were considered
BFTBP: #reads=58051 , insert_mean=2000, insert_stdev=666
BFTDP: #reads=10409 , insert_mean=2000, insert_stdev=666
4. The reads have been retrimmed using veraTrim (-T 10 -M 100 -E 500)
5. runCA-OBT.pl has been used to assemble all the reads
location: 2007_0724_WGA-default/
=>160 scaff, 163 contigs, 23X coverage
6. The library sizes were updates using the WGA estimates
BFTBP: insert_mean=2690.042, insert_stdev=643.126
BFTDP: insert_mean=3675.914, insert_stdev=1225
7. The WGA was aligned to the reference using nucmer; one rearrangement, one
deletion and several SNP's were noticed
8. The reads were assembled using AMOScmp (default parameters)
location: 2007_0724_AMOSCMP-default/
=> 1 scaffold, 22 contigs
2 missoriented read pile regions were noticed
9. The assembly was aligned to itself; 950 bp inverted repeats were identified as
flanking the problem regions; the coordinates are:
16336-21562 (5 KB)
167086-184936 (17 KB)
10. The 2 regions were flipped ; the new reference is called NC_008369.2
11. Several small contig (step 8) read clear ranges have been extend to their OBT
trimming points
12. AMOScmp was rerun using more relaxed parameters:
nucmer MINCLUSTER=30
casm-layout MAXTRIM=50
location: 2007_0731_AMOSCMP-veraTrim-updateDst-relaxed-updateClr-fixRef2->best
=> 1 scaffold, 8 contigs
Francisella_tularensis.qc
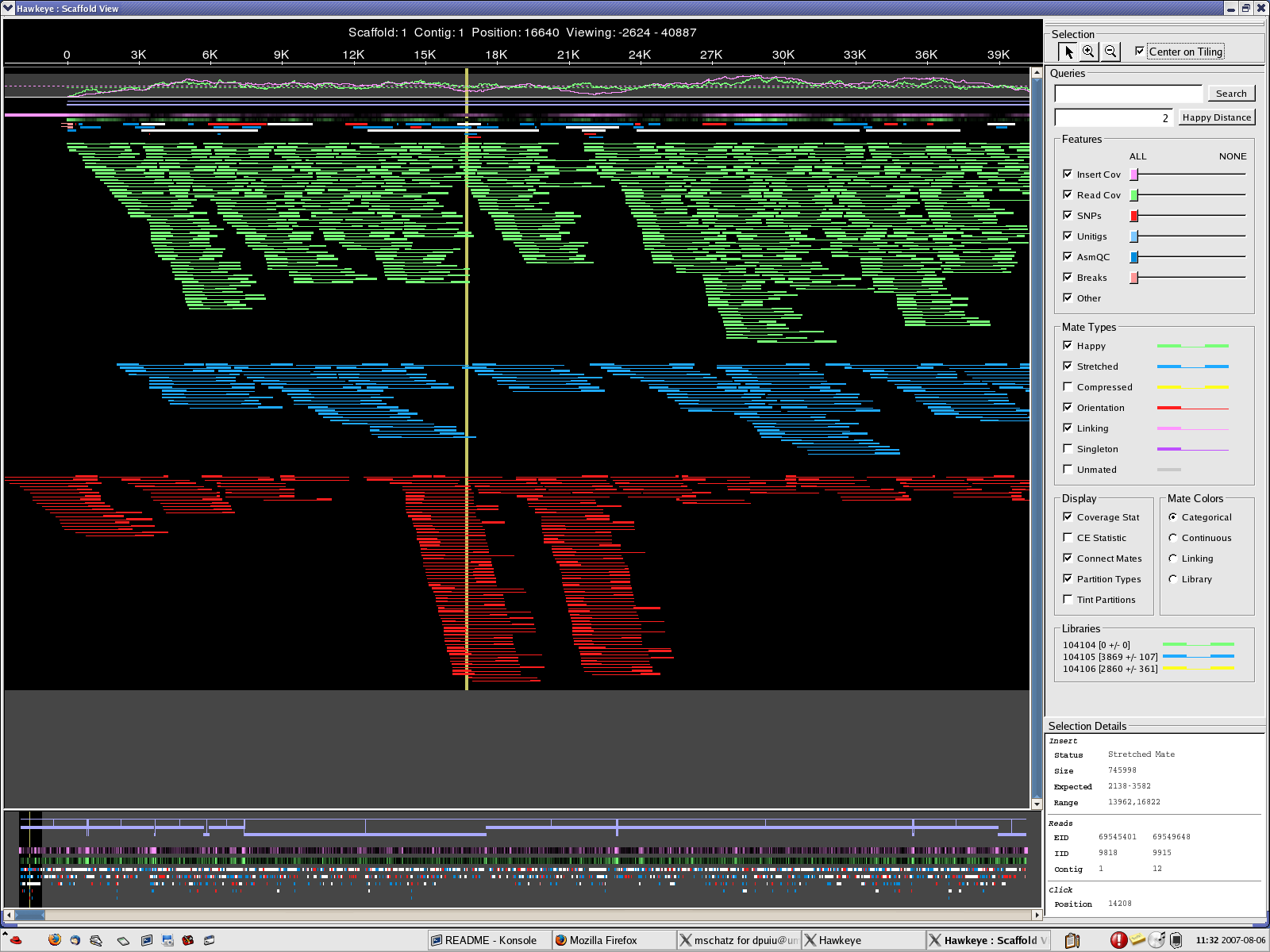{kind=link}
{kind=link}
Location:
/fs/szasmg/Bacteria/F_tularensis_holarctica_OSU18/ /fs/szasmg/Bacteria/F_tularensis_holarctica_OSU18/best # Final assembly
Annotation
Location:
/fs/szdata/ncbi/genomes/Bacteria/Francisella_tularensis_holarctica_OSU18 NC_008369.faa : translated CDS NC_008369.ffn : CDS positions & sequnce NC_008369.ptt : protein table NC_008369.rnt : RNA table
1555 genes were aligned to the new reference using nucmer
most genes had unique alignments some had multiple alignments 2 had partial alignments (part in the gap) 3 had no alignments File location: /fs/szasmg/Bacteria/F_tularensis_holarctica_OSU18/best/NC_008369.ptt # remapped gene table /fs/szasmg/Bacteria/F_tularensis_holarctica_OSU18/best/NC_008369.rnt # remapped RNA table
Kunmi's link:
http://cbcbwww01.umiacs.umd.edu/cgi-bin/sybil_demo/shared/show_protein.cgi?site=demo&id=7325110
NCBI TPA submission
- Inverted regions:
16336-21562 167086-184936
- Mate evidence:
17 inv1.3.tab.tis 25 inv1.5.tab.tis 13 inv2.3.tab.tis 25 inv2.5.tab.tis 160 inv.all.tis
- Used Seqin
- File: /fs/ftp-cbcb/pub/data/F_tularensis_holarctica_OSU18/CP000437.2.sqn (generated by Seqin)
- Assigned TPA accession number (09/23/08): BK006741
- File: /fs/ftp-cbcb/pub/data/F_tularensis_holarctica_OSU18/BK006741.fna
- BK006741
- NCBI Whole Genome Shotgun Sequence Submissions
- Locus-tag prefix AYI
- Genome Project Registration
- GenomeProject ID #32025
NCBI AA submission
- AA submission prepared in
/fs/szasmg/Bacteria/F_tularensis_holarctica_OSU18/2007_0731_AMOSCMP-veraTrim-updateDst-relaxed-updateClr-fixRef2/AA