Bos taurus 3.0
Sequence
The genome of the domestic cow, Bos taurus, was sequenced using a mixture of hierarchical and whole-genome shotgun sequencing methods.
Read download
- All reads were downloaded from the NCBI Trace Archive (TA) ftp: ftp://ftp.ncbi.nih.gov/pub/TraceDB/bos_taurus/
- There were 37,829,394 reads organized into 91 volumes
- 36,820,485 WGS, SHOTGUN, CLONEEND & FINISHING reads
- 36,170,352 quality reads
- 650,133 quality-less reads
- 1,008,909 EST & PCR reads
- 36,820,485 WGS, SHOTGUN, CLONEEND & FINISHING reads
- 25,312 read libraries
Sequencing centers
- Most reads were sequenced by the Baylor College of Medicine
TRACE_COUNT CENTER_NAME
1 35629020 BCM Baylor College of Medicine
2 737900 NISC NIH Intramural Sequencing Center
3 652614 BCCAGSC British Columbia Cancer Agency Genome Sciences Center
4 378871 MARC USDA, ARS, US Meat Animal Research Center
5 114753 UIUC University of Illinois at Urbana-Champaign
6 107367 BARC USDA, ARS, Beltsville Agricultural Research Center
7 65171 TIGR The Institute for Genome Research
8 53556 GSC Genoscope
9 43033 CENARGEN Embrapa Genetic Resources and Biotechnology
10 18623 SC The Sanger Center
11 15301 UOKNOR University of Oklahoma Norman Campus, Advanced Center for Genome Technology
12 10651 TIGR_JCVIJTC The Institute for Genomic Research, Traces generated at JCVIJTC
13 2485 UIACBCB University of Iowa Center for Bioinformatics and Computation Biology (UIACBCB)
14 49 WUGSC Washington University, Genome Sequencing Center
37829394 total total
Trace counts
TRACE_COUNT CENTER_NAME TRACE_TYPE_CODE
1 24863599 BCM* WGS
2 10748529 BCM* SHOTGUN
3 737900 NISC SHOTGUN
4 125597 BCCAGSC CLONEEND
5 114753 UIUC CLONEEND
6 65171 TIGR CLONEEND
7 53556 GSC CLONEEND
8 26246 CENARGEN WGS
9 25454 BARC CLONEEND
10 16892 BCM* CLONEEND
11 16787 CENARGEN CLONEEND
12 15150 UOKNOR SHOTGUN
13 10651 TIGR_JCVIJTC CLONEEND
14 151 UOKNOR FINISHING
15 49 WUGSC CLONEEND
36820485 total
16 527017 BCCAGSC EST
17 207204 MARC EST
18 171667 MARC PCR
19 81913 BARC EST
20 18623 SC EST
21 2485 UIACBCB EST
1008909 total
Data processing
Data issues
Issues:
- Qualities
- 650,133 reads don't have quality values and can't be reliably trimmed
- Libraries
- There are totally 25,312 libraries
- Very fragmented especially the SHOTGUN and CLONEEND ones; can't be accurately re-estimated by the assembler
- Clear ranges
- Many traces are missing vector trimming coordinates (CLV=CLIP_VECTOR_LEFT..CLIP_VECTOR_RIGHT) or don't contain 3' trimming information (CLIP_VECTOR_RIGHT==0)
- The read CLV's are need by the Celera Assembler overlap based trimming module (OBT) as input
- Solution: identify the sequencing vector & linker sequences for each library and re-trim the reads
Identify linkers
For each library identify linker sequences:
- Separate forward/reverse reads
- Identify most frequent kmers (8mers,24mers)
- Check if kmers a overrepresented
- Verify if the most frequent 8mer is present in the top 10 most frequent 24mers
- Align 24mers (extend them by a few bp) => linker
Identify vectors
For each library identify vector sequences:
- Align linkers to the opposite strand sequences (nucmer -l 12 -c 24 -r)
- Extract the subsequences following to linker (50..150bp)
- Align the subsequences; if they align we've probably identified the vector
- Identify the vector name/id by alignment to the UniVec database (nucmer -l 12 -c 24)
- Check if the forward/reverse vector(s) are the same : we should find a common vector sequence; the UniVec alignments should be adjacent
- create the Lucy vector & splice files that contain the linker+vector sequences
Trimming
- Run Lucy on quality reads
- Get CLV statistics: depending on the library, the Lucy CLV is 20bp+ shorter than the original CLV
- Trim reads according to Lucy output CLV
- Align Lucy trimmed reads to linker,vector,splice site & UniVec (there should be no alignments)
- Method worked on BCM & NISC libraries (~ 98% of the reads)
- For the other reads use the factory clipping points
BCM reads
- linker:
>J01636.linker.fwd 27bp TCGAGTTCGACTGCAAGTAGTTCATCA >J01636.linker.rev 27bp CTAATCAGATGGTACAGTAGTTCATCA
- vector: J01636 E.coli lactose operon with lacI, lacZ, lacY and lacA genes (7477 bp)
- avg(original CLV) - avg(Lucy CLV)> 20bp (1015 vs 973 in quality WGS reads , ...)
NISC reads
- linker:
>NGB00080.linker.fwd 24bp TATCATCGCCACTGTGGTGGAATT >NGB00080.linker.rev 26bp GCTGAAGCTCCATGTGGTGGAATTCC
- vector NGB00080 (pOTW13 with linkers)
- avg(original CLV) - avg(Lucy CLV)> 20bp (771 vs 747)
Preliminary assembly
- Assembly version: wgs-5.2
- Use only quality reads
- Set read CLV to Lucy CLV or original CLV
- Set non random flag = 1 on all reads except for WGS ones
- Set obtMerThreshold = 200 (default 1000)
- Set doOBT = 1
Input
Reads=35,348,776 # WGS, SHOTGUN, CLONEEND & FINISHING quality reads Libraries=25,312 # mostly SHOTGUN and BARC.CLONEEND
Output
TotalScaffolds=66,141 MaxBasesInScaffolds=26,048,998 MeanBasesInScaffolds=40,861 TotalContigsInScaffolds=120,461 MaxContigLength=627,911 MeanContigLength=22,436 TotalDegenContigs=269,031 MaxDegenContig=33,824 SingletonReads=3,721,123
DeletedReads=421,379 (too short or zero CLR)
Preliminary assembly processing
Read clear ranges
- Quality reads: extract OBT CLR from gatekeeper store
- Qualityless reads:
- Align them to contigs (no degenerates) : nucmer -l 50 -c 200 -b 10 -g 5 -d 0.05
- Set CLR to the maximum alignment coordinates or 50..min(len,600)
- Reduce CLR if there are multiple N's or low complexity regions in the read
Contaminant search
Databases:
- Ecoli : 22 completed genomes + plasmids
- UniVec_Core 1,348 sequences : mostly cloning vectors & primers, avg 250bp long
- OtherVec: 100 other vector sequences (mostly complete), identified by aligning UMD2.0 contaminants to GenBank
- bos_taurus UMD2.0 contaminant : 4,813 whole contigs and 30,329 partial contigs identified by NCBI as contamination in UMD2.0; many partial contigs contained cow sequences as well
- Databases FASTA files:
/nfshomes/dpuiu/db/Ecoli.all /nfshomes/dpuiu/db/UniVec_Core /nfshomes/dpuiu/db/OtherVec /nfshomes/dpuiu/db/bos_taurus.UMD2.contaminant.fasta
Alignment parameters:
nucmer -maxmatch -l 40 -c 100 -b 10 -g 5 -d 0.05
Contig/degenerate counts:
- 2,951/1,266 aligned to Ecoli
- 5,387/1,908 aligned to UniVec_Core
- 5,657/1,963 aligned to OtherVec
Read/mate counts: TO BE DELETED
- 40,699/22,607 in contaminated regions
Library estimates
- Some library estimates are complete wrong
Example: BCM.SHOTGUN libraries listed as long (180Kbp mean) are all short (2-6Kbp mean)
- Extract library insert estimates; merge libraries sequenced by same center that have similar mean/std : 25,312 libs => 344 libs
- Assign new library ids; assign average means & stdevs to the libraries
Final assembly
- Assembly version: wgs-5.2
- Use all traces
- Set read CLR to:
- Quality reads: OBT CLR
- Qualityless reads: alignment coordinates or 50..min(len,600)
- Set nonRandom flag = 1 on all reads except for WGS reads
- Set deleted flag = 1 on all reads deleted by OBT in the preliminary assembly
- Set obtMerThreshold = 200 (default 1000)
- Set doOBT = 0 (reads have been already trimmed)
Input
Reads=35,973,728 # WGS, SHOTGUN, CLONEEND & FINISHING with and without qualities Libraries=344
Output
TotalScaffolds=39,978 TotalContigsInScaffolds=90,135 MeanBasesInScaffolds=66,947 MaxBasesInScaffolds=3,3907,885 TotalContigsInScaffolds=90,135 MeanContigLength=29,693 MaxContigLength=1,160,130 TotalDegenContigs=251,413 MaxDegenContig=39,964 SingletonReads=3,634,305(10.24%)
Final assembly processing
Contaminant search
- Use same databases and alignment parameters as in preliminary assembly processing
- Delete full contaminants & trim partial contaminants
Delete summary:
- 65 Acinetobacter ctgs
- 91 other contaminant ctgs <2Kbp
- Total: 156 ctgs, 152 scf, 4105 reads
Trim summary:
- 12 contigs >=2Kbp , 44 reads
Marker mapping
- 126,013 total markers
- Avg distance between markers is 25Kbp; marker position error is 50Kbp
- Markers were aligned to all contigs/degenerates
- Best alignments with %IDY>90 & %Matched>85 were identified
- 107,271 markers align to 31,407 ctg & 2,640 scf
- 552 scf have markers from multiple chromosomes
- 212 scf have multiple markers from multiple chromosomes
- 38 scf have multiple adjacent markers from multiple chromosomes: MIGHT BE MISASSEMBLED
- 628 markers align to 562 degenerates
Scaffold/contig breaking
- Analyze 38 scf that have multiple adjacent markers from multiple chromosomes
- Compute coverage in the suspicious region (between different chromosome markers):
- read cvg
- mate ctg: good, bad
- Break ctg/scf unless the region has "high read cvg" , "high good mate cvg" , "low bad mate cvg"
- Break summary:
- 14 scaffolds
- 15 breaks : 8 on the same contig , 3 on adjacent contigs , 4 on non adjacent contigs
Assignment to chromosomes
Markers
- 2640 scaffolds and 562 degenerates have markers
- Assignment to chromosomes: use best alignment & majority rule
- Position:
- Filter out outliers according to position on chromosome & scaffold (interquartile range method)
- Compute the average position on chromosome of the markers
- Orientation:
- use LeastSequareFit method : if slope is positive => forward; if slope is negative => reverse
- if only 1 markers/scaffolds => direction=unknown (0)
Human synteny
- Align all scaffolds/degenerates to the 24 Human chromosomes; filter all alignments longer than 200bp
nucmer -mum -l 12 -c 30 -g 1000 delta-filter -q -l 200
- 9,914 scaffolds and 16,527 degenerates align to Human chromosomes; most alignments are short, just over 200bp
Combine Human synteny & Marker data
- 1,908 scaffolds and 118 degenerates both align to human and contain markers
- 10,790 scaffolds and 16,590 degenerates align to human or contain markers
- Try to infer the position/orientation on the chromosomes for the scaffolds/degenerates that align to human but contain no markers
- Iteratively:
- Find 2 adjacent scaffolds (preferably on left & right side) which both align to human, contain markers and placements agree (chromosome, position, direction)
- Otherwise, find 1 adjacent scaffolds which both aligns to human, and contains markers
- Extrapolate the position/orientation of the "unplaced" sequence based on its neighbor(s)
- Sort the scaffolds/degenerates based on chromosome positions, identify incorrect markers & alignments, remove them from the input data and repeat the process
By linking information
- Once scaffolds/degenerates were assign to chromosome use mate pair information to refine placements
- Identify unplaced scaffolds/degenerates linked to placed scaffolds/degenerates and fit them into gaps
Comparison to UMD2.0
Alignment parameters:
nucmer -mum -l 200 -c 1000
Haplotype search
Daniela:
- Place scf/deg on Chr
- Align each pair X,Y (len(X)<len(Y))of adjacent/overlapping scf/deg : nucmer -mum -l 40 -c 250 ( => avg 96 %id)
- compute (X,Y) cvg
- identify the X regions which had no alignments to Y; if the length of these regions were less than 2K bp => X is a variant
Guillaume:
- there is a contig Y larger than X and, X and Y are placed on top of each other (with some play allowed)
- there are a high quality sequence alignment such that: at least 200 bases out of the first 400 bases of X align with Y AND at least 200 bases out of the last 400 bases of X align with Y.
- In other word, the ends of X have to align well with Y, but the middle can be significantly different.
Files:
/fs/szasmg3/bos_taurus/UMD_Freeze3.0/contigs.haplotype-variants.fa.gz # 40611 haplotype-variants sequences /fs/szasmg3/bos_taurus/UMD_Freeze3.0/contigs.haplotype-variants.ids # 40611 haplotype-variants sequence ids /fs/szasmg3/bos_taurus/UMD_Freeze3.0/contigs.haplotype-variants.pairs # 40300 pairs (haplotype-variants & reference sequences) /fs/szasmg3/bos_taurus/UMD_Freeze3.0/contigs.haplotype-variants.pairs.delta # 39665 alignment pairs (haplotype-variant is the query) /fs/szasmg3/bos_taurus/UMD_Freeze3.0/contigs.haplotype-variants.pairs.cvg # 39665 coverage pairs (ref=col 1 ; haplotype-variant=col 5)
Summary:
. elem <=0 >0 min max mean med n50 sum ctg+deg 40611 0 40611 263 97877 1476 1205 1372 59958728 ctg 29452 0 29452 471 97877 1631 1297 1469 48039280 deg 11159 0 11159 263 12208 1068 979 1006 11919448
Other Files
/scratch1/bos_taurus/Assembly/2009_0312_CA/scf_placements/UMD_Freeze3.0/ 39864 contigs.haplotype-variants.daniela.pairs 443 contigs.haplotype-variants.guillaume.pairs 436 contigs.haplotype-variants.guillaume.pairs.orig 334 contigs.haplotype-variants.ids.missing
Issues:
. elem <=0 >0 min max mean med n50 sum missing 339 0 339 462 97877 1613 1011 1204 547135 mislabeled 6 0 6 2973 97877 31271 8275 97877 187628
Mislabeled haplotypes:
Chr begin end Pos W ctg 1 len dir Chr2 131428091 131475109 5387 W 7180001925346 1 47019 + Chr3 11270384 11278308 637 W 7180002020315 1 7925 - Chr12 9183395 9281271 429 W 7180002024890 1 97877 + Chr14 34404395 34412669 3579 W 7180002021388 1 8275 - Chr15 50865607 50889165 2531 W 7180002015261 1 23559 - ChrU 8589989 8592961 5791 W 7180002026074 1 2973 +
Chromosome mapping
Assembly Summary
...
Hs vs Bt
- Goal: find all syntenic regions longer than a certain % of the Cow/Human genome
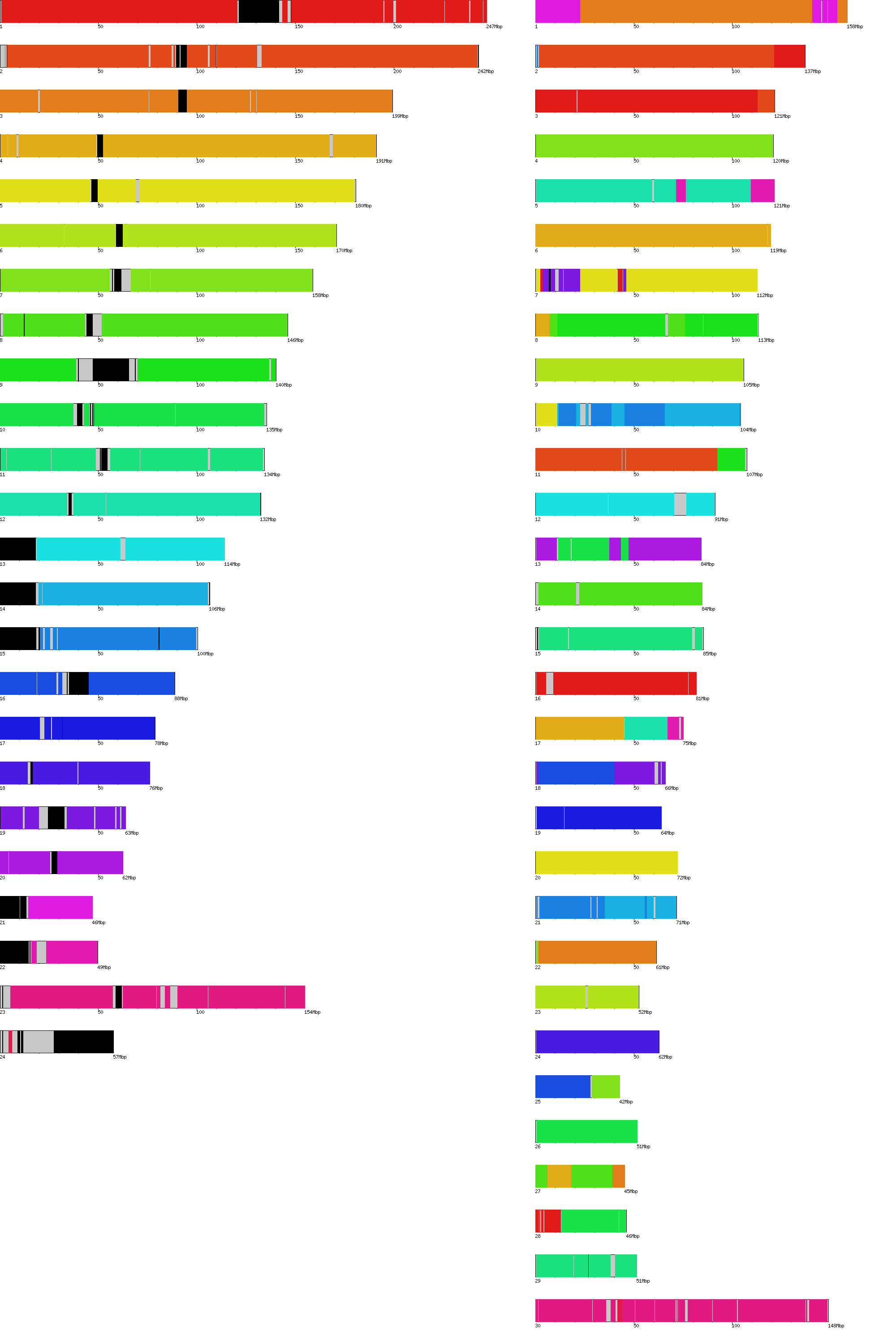
- Chromosome counts (include gaps)
. elem min q1 q2 q3 max mean n50 sum(all) sum(no gaps) human 24 46944323 78774742 134452384 170899992 247249719 128350811 154913754 3,080,419,480 2,858,012,910 cow 31 9828056 61435874 84240350 113384836 158337067 86152724 105708250 2,670,734,461 2,649,997,198
- Gap counts
. elem min q1 q2 q3 max mean n50 sum human 290 100 35000 47000 90000 30000000 766919 17918000 222,406,570 => 7.2% gaps cow 72454 1 99 99 248 1074158 286 698 20,737,263 => 0.7% gaps
- nucmer params: -l 12 -c 65 -g 1000 -b 1000
- delta-filter -l 200
- 24 * 30 = 720 alignments (except for BtChrU)
- Alignment stats (filter-q)
. elem min q1 q2 q3 max mean n50 sum len 392789 11 440 749 1244 34597 1015 1376 398713561 %id 392789 30.06 74.72 77.89 81.61 100.00 78 78 .
- Alignments counts
>=200 >=2000 >=5000 HsChr-BtChr.delta 532,866 39,663 3,570 HsChr-BtChr.filter-1.delta 392,789 38,185 3,560
- 54 chr sets have at least one 5K alignments
- HsChrX-BtChrX.png
- HsChrX-BtChrX.filter-2K.png
- HsChrX-BtChrX.filter-5K.png
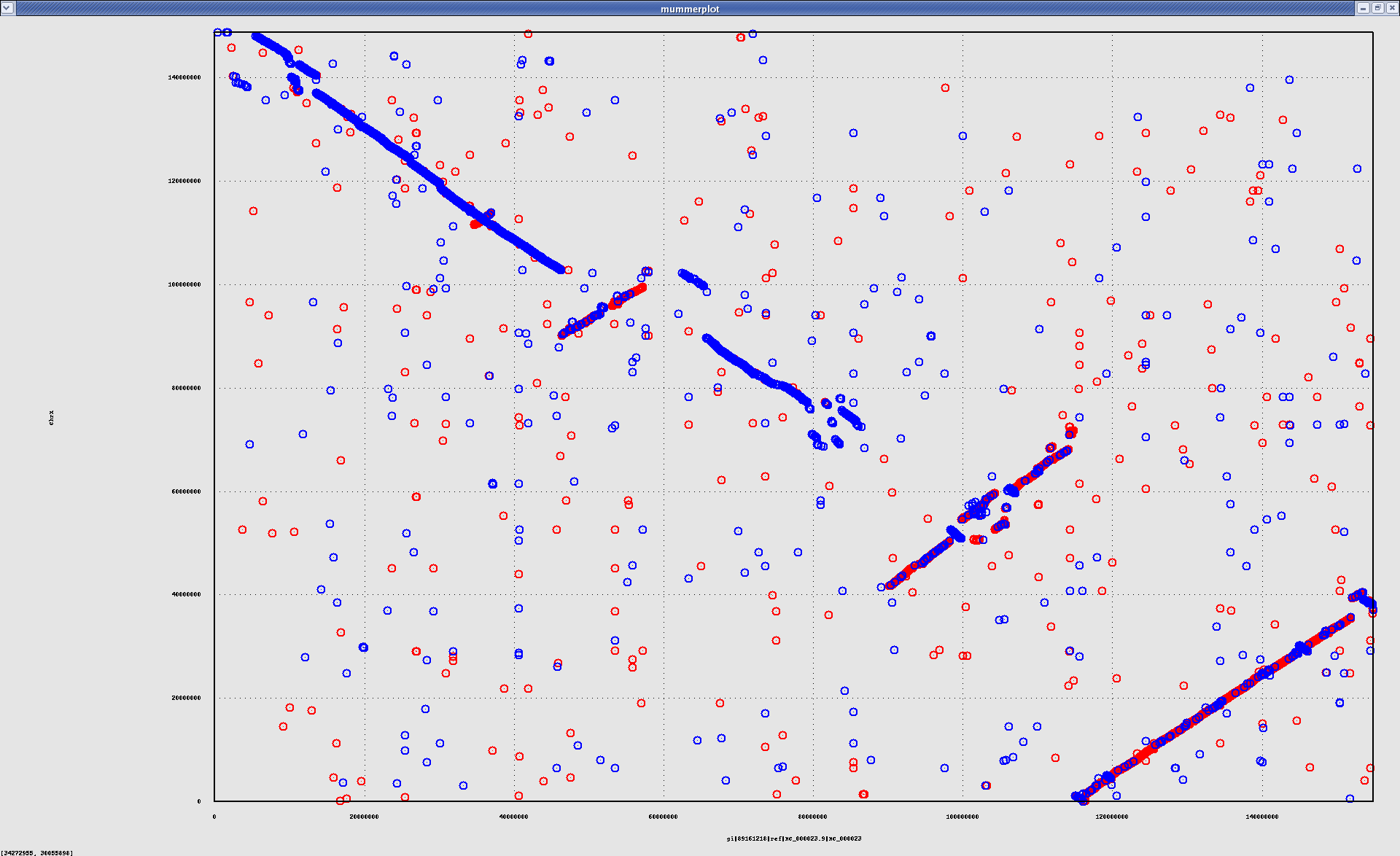{kind=link}
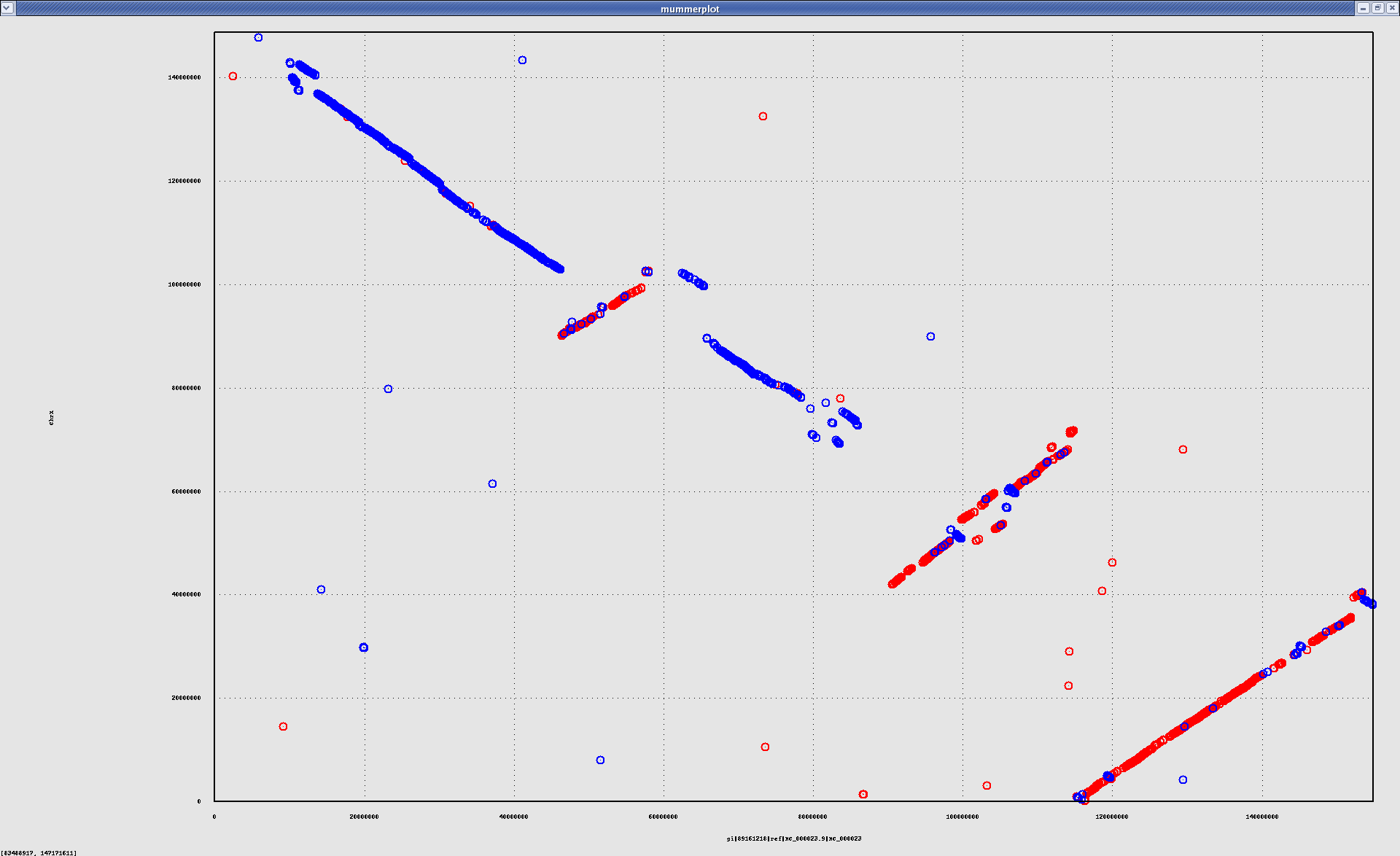{kind=link}
{kind=link}
- Filter and merge alignments
cat HsChr-BtChr.filter-1.delta | ~/bin/shrinkIds.pl | ~/bin/DELTA/delta2anc.pl | sed 's/NC_//' | sort -nk1 -nk5 | sed 's/^/NC_/' >! HsChr-BtChr.anc
cat HsChr-BtChr.anc | ~/bin/DELTA/filter-anc.pl -p 0.2 >! HsChr-BtChr.filter.anc
cat HsChr-BtChr.filter.anc | ~/bin/DELTA/merge-anc.pl >! HsChr-BtChr.merge.anc
cat HsChr-BtChr.merge.anc | ~/bin/DELTA/anc2delta.pl >! HsChr-BtChr.merge.delta
392789 HsChr-BtChr.anc
368848 HsChr-BtChr.filter.anc
380 HsChr-BtChr.merge.anc : 380 syntenic regions !!!
- Alignment lengths
. elem min q1 q2 q3 max mean n50 sum filter-1 392789 11 440 749 1244 34597 1015 1376 398713561 => 13.9% of finished human covered merge 323 191406 1363213 3413785 10424392 86757102 8304710 19750702 2682421368 => 93.8% of finished human covered
- Coverage
cat HsChr-BtChr.merge.delta | ~/bin/delta2cvg.pl -m 2 | getSummary.pl -i 4 -t 2+cvg . elem min q1 q2 q3 max mean n50 sum merge(2+cvg) 15 174 222 341 541 1651 455 541 6829 !!! only 6K in overlapping regions
- Plot
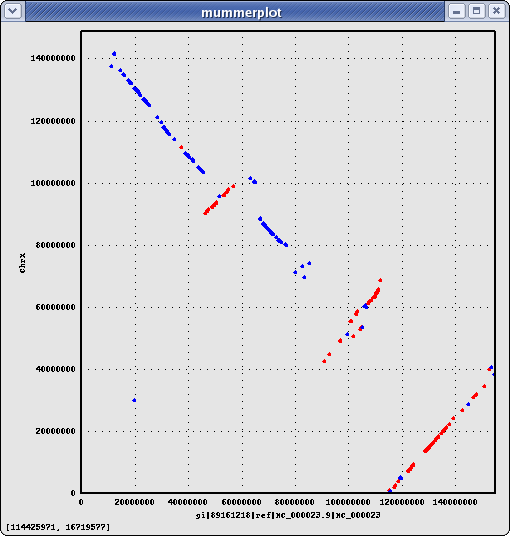
cat HsChr-BtChr.merge.delta | ~/bin/DELTA/mummerplot.pl
cat HsChr-BtChr.merge.anc | sed 's/NC_00000//' | sed 's/NC_0000//' | sed 's/Chr//' | sed 's/X/30/' | p '@F[6,7]=@F[7,6] if($F[6]>$F[7]); print join " ", @F[0,4,5,1,6,7]; print "\n";' > HsChr-BtChr.merge.map ~/bin/map-draw.pl -rl HsChr.len -ql BtChr.len -rg HsChr.gaps -qg BtChr.gaps HsChr-BtChr.merge.map >! HsChr-BtChr.merge.png
{kind=link}
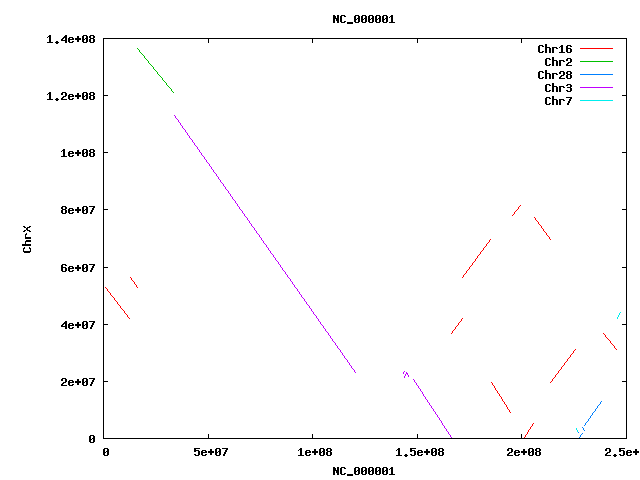{kind=link}
{kind=link}
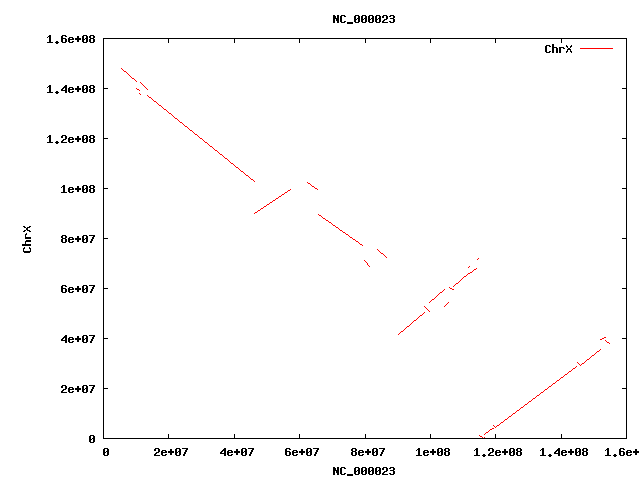{kind=link}
Submission
Issues
3 tbl2asn.3Nucleotides.ids # 3 deg 13 tbl2asn.InternalNs.ids 7 tbl2asn.TerminalNs.ids # 3 deg
NCBI contaminant search
Abbr. Screen type Total Exclude Mask/trim x_dist Not chordata 15 8 x_mito Mitochondrial 75 74 # +2 degenerates found later on x_rel Primates, Glires 61 18 x_vec Vector 72 17 52
- 115 sequences to exclude (with apparent source)
- 56 sequences with locations to mask/trim (with apparent source)
- More mito contaminants that got deleted:
contig 7180001836672 (941 bp) Chr4 contig 7180001872458 (1216 bp) Chr7
USDA validation
- contigs where the USDA markers are:
#ctgid chr chrStart chrEnd chrDir gap gapLen scfid scfStart scfEnd scfDir 7180001851853 Chr4 14880886 14905494 f N 262 7180002041025 0 24609 f # UMD2.ChrX 7180001851854 Chr4 14905757 14975933 f N 89 7180002041025 25050 95227 f # UMD2.ChrX 7180001851855 Chr4 14976023 15035183 f U 100 7180002041025 95247 154408 f # UMD2.ChrX 7180001851862 Chr4 15097101 15112881 f N 277 7180002041025 214346 230127 f # UMD2.ChrU 7180001851863 Chr4 15113159 15217082 f U 100 7180002041025 230471 334395 f # UMD2.ChrU 7180001851868 Chr4 15315244 15402777 f U 100 7180002041025 428933 516467 f # UMD2.ChrU 7180001851869 Chr4 15402878 15481630 f U 100 7180002041025 516487 595240 f # UMD2.ChrU 7180001851870 Chr4 15483895 15545907 f N 1420 7180002041025 595301 657314 f # UMD2.ChrU 7180001851877 Chr4 15750682 15829262 f N 113 7180002041025 872729 951310 f # UMD2.Chr4 7180001851878 Chr4 15829376 15899492 f U 100 7180002041025 951330 1021447 f # UMD2.Chr4 7180001851883 Chr4 16063597 16181903 f U 100 7180002041025 1185217 1303524 f # UMD2.Chr4 7180002017095 Chr4 46537074 46586634 f N 106 7180002041269 4370630 4420191 f # UMD2.ChrX ; 49,561bp ctg (ctg 60 out of 68 in the scaffold)
- scf7180002041025 & scf7180002041269 should go on Chr X
scf7180002041025 1504672 (1.5Mbp) 40 ctgs scf7180002041269 5135095 (5.1Mbp) 68 ctgs
scf7180002041025
- aligns to human ChrX
- has cow Chr4 markers
#scfid chr #markers 7180002041025 4 6
- 4 more scaffolds that align inside of it got placed on Chr4
- ChrX synteny break
#id HS-ref #alignments slope begin end len BT-ref #markers slope begin end # ctgs #before: scf 7180002041067 #7180002041067 23 218 1.2073 110927695 111673606 745911 30 11 0.8025 68190660 68936571 7180002041025 23 170 -0.7174 114577935 116082607 1504672 4 6 0.9532 15769548 17274220 40 7180002035944 23 3 -0.0005 115565458 115615980 50522 . . . . . 6 # 1 link to 7180002041025, 1 link to 7180002041025(Chr14) 7180002038855 23 1 -1.0258 115614308 115620479 6171 . . . . . 1 7180001954604 23 2 0 115819587 115820534 947 . . . . . 1 (deg) 7180002066413 23 1 0.996 115849570 115850802 1232 . . . . . 1 # 1 link to 7180002041025 , 4 links to 7180002040813(Chr23) #after: scf 718000204081 (ctg 7180001725840..7180001725857) #7180002040813 23 448 0.9565 116445525 117676925 1231400 30 23 0.8741 208791 1440191
- 7180002041025 has 2 mate links to 7180002040813 & zero links to 7180002041067
#read1 read2 type scf1 begin1 end1 dir1 scf2 begin2 end1 dir2 448279509 581499140 diffScaffold 7180002041025 14677 15288 r 7180002040813 156247 156894 r # 581499140(UIUC CLONEEND) 395754360 395754351 diffScaffold 7180002041025 86506 87216 r 7180002040813 21906 22622 r # 395754351(TIGR CLONEEND)
MOVE 5 SCF (48 CTG + 1 DEG = 49 CTG/DEG) FROM Chr4 TO ChrX: 7180002041025 ...
BEFORE SCF 7180002040813 (reads 395754351,581499140) : 1st scf in Chr30, forward
scf7180002041269
- has 1 marker from cow ChrX
#scfid chr #markers 7180002041269 4 211 7180002041269 2 2 7180002041269 27 1 7180002041269 X 1
#Marker Chr_BTA Pos(Kbp) CI_Pos_from CI_Pos_to UMD_Ctg_Pos Match_Len %IDY %Matched UMD_Ctg_name BZ868101 30 117730101 117682601 117777601 32868 607 99.84 100.00 7180002017095
7180002017095(49561bp) --(8)--> 7180001836903(5012bp) --(10)--> 7180002017096(14286bp) --(3)--> 7180002000237(5737bp) --(7)--> 7180002017097(67824bp,Chr4)
--(4)--> 7180001765615(2394bp)
- 1Kbp+ alignments:
26294 27384 | 34011599 34012646 | 1091 1048 | 74.49 | 49561 154913754 | 2.20 0.00 | 7180002017095 gi|89161218|ref|NC_000023.9|NC_000023 10631 12096 | 34107297 34108751 | 1466 1455 | 82.36 | 14286 154913754 | 10.26 0.00 | 7180002017096 gi|89161218|ref|NC_000023.9|NC_000023
BREAK SCF 7180002017096
MOVE 3 CONTIGS FROM Chr4 TO ChrX: 7180002017095, 7180001836903, 7180002017096 ( align to human ChrX)
BETWEEN ctg 7180002005166,7180002013375 ; forward
#scfid HS-ref #align slope HS-beg HS-end scflen #ctgid 7180002035547 23 7 -0.6625 33257979 33286095 28116 7180002005166|7180002013375 (2ctg scf) ... 7180002040082 23 4 1.4432 34269717 34305189 35472 7180002021537 (1ctg scf)
#BT agp ChrX 113665631 113671433 13435 W 7180002005166 1 5803 + ... ChrX 113671746 113693999 13437 W 7180002013375 1 22254 +
- MOVE 50 CTGS from Chr4 to ChrX (2 variants 7180001912167 & 7180001954604 skipped)
1,532,083 bp in ctg & gaps 1,513,442 bp in ctg 18,641 bp in gaps
Before After Chr4 122,361,782(2,761 ctg) 120,829,699(2,711 ctg) ChrX 147,291,816(8,628 ctg) 148,823,899(8,678 ctg)
Wrong Haplotypes
Dpuiu 15:16, 24 August 2009 (EDT)
Mislabeled haplotypes:
Chr begin end Pos W ctg 1 len dir Chr2 131428091 131475109 5387 W 7180001925346 1 47019 + Chr3 11270384 11278308 637 W 7180002020315 1 7925 - Chr12 9183395 9281271 429 W 7180002024890 1 97877 + Chr14 34404395 34412669 3579 W 7180002021388 1 8275 - Chr15 50865607 50889165 2531 W 7180002015261 1 23559 - ChrU 8589989 8592961 5791 W 7180002026074 1 2973 +
New files:
/scratch1/bos_taurus/Assembly/2009_0312_CA/scf_placements/UMD_Freeze3.1
Mislabeled haplotypes (added back to the assembly)
Chr begin end Pos W ctg 1 len dir Chr2 131428091 131475109 5387 W 7180001925346 1 47019 + Chr3 11270384 11278308 637 W 7180002020315 1 7925 - Chr12 9183395 9281271 429 W 7180002024890 1 97877 + Chr14 34404395 34412669 3579 W 7180002021388 1 8275 - Chr15 50865607 50889165 2531 W 7180002015261 1 23559 - ChrU 8589989 8592961 5791 W 7180002026074 1 2973 +
NCBI links
--Dpuiu 15:54, 9 September 2009 (EDT)
- We have released the 02 version of your WGS project plus the scaffolds and chromosomes:
GPID Orgname WGS number -------------------------------- 32899 Bos taurus DAAA00000000
GK000001-GK000030 = chromosomes (made from the scaffolds) GJ011756-GJ060422 = 48,667 scaffolds
- The chromosomes are updates to the pre-existing accession numbers, so they are now the .2 version.
- GK000001-GK000030 are chr1-29 and chrX, respectively.
- We added the last 6 contigs to the WGS project, so it has 75770 contigs.
- There will be the usual indexing delay before text searches and the hyperlinks function correctly.
GK000001-GK000030 chromosomes (same accession numbers) GJ060423-GJ063645 placed scaffolds (new accession numbers) GJ057137-GJ060422 unplaced scaffolds (same accession numbers)
CBCB links
http://www.cbcb.umd.edu/research/production_assembly.shtml ftp://ftp.cbcb.umd.edu/pub/data/assembly/Bos_taurus/Bos_taurus_UMD_3.0 -> /fs/ftp-cbcb/pub/data/Bos_taurus/Bos_taurus_UMD_3.0 ftp://ftp.cbcb.umd.edu/pub/data/assembly/Bos_taurus/Bos_taurus_UMD_3.0a -> /fs/ftp-cbcb/pub/data/Bos_taurus/Bos_taurus_UMD_3.0a (alpha release) ftp://ftp.cbcb.umd.edu/pub/data/assembly/Bos_taurus/Bos_taurus_UMD_3.1 -> /fs/ftp-cbcb/pub/data/Bos_taurus/Bos_taurus_UMD_3.1 (AGP gap specification change)
/fs/szasmg3/bos_taurus/UMD_Freeze3.0 # AGP & Chr Seqs /fs/szasmg3/bos_taurus/UMD_Freeze3.1 # new AGP /fs/szasmg3/bos_taurus/UMD_Freeze3.1.NCBI # NCBI Chr Seqs (NCBI ids)
Marker mapping
- About 75% of the MARC markers and 87% of the ILTX seem to agree (chromosome number and approximate position).
BTAU4.2_UMD3.1.MARC.txt BTAU4.2_UMD3.1.ILTX.txt
- MARC markers mapping summary:
total markers: 1384 same chromosome: 1047 different chromosome: 17 BTAU4.2 only: 355 UMD3.1 only: 13
- ILTX markers mapping summary:
total markers: 3396 same chromosome: 2988 different chromosome: 169 BTAU4.2 only: 327 UMD3.1 only: 0
Files:
BTAU4.2 vs UMD3.1 /fs/szasmg3/bos_taurus/markers/BTAU4.2_UMD3.1.MARC.txt /fs/szasmg3/bos_taurus/markers/BTAU4.2_UMD3.1.ILTX.txt
MARC makers /fs/szasmg3/bos_taurus/markers/9913.MARC.txt : marker positions on BTAU4.2 (ftp file) /fs/szasmg3/bos_taurus/markers/UMD3.1.MARC.txt : marker positions on UMD3.1 /fs/szasmg3/bos_taurus/markers/MARC.txt : marker ids /fs/szasmg3/bos_taurus/markers/MARC.fwd.seq : marker forward sequences /fs/szasmg3/bos_taurus/markers/MARC.rev.seq : marker reverse sequences
ILTX makers: /fs/szasmg3/bos_taurus/markers/9913.ILTX.txt : marker positions on BTAU4.2 (ftp file) /fs/szasmg3/bos_taurus/markers/UMD3.1.ILTX.txt : marker positions on UMD3.1 /fs/szasmg3/bos_taurus/markers/ILTX.txt : marker ids /fs/szasmg3/bos_taurus/markers/ILTX.fwd.seq : marker forward sequences /fs/szasmg3/bos_taurus/markers/ILTX.rev.seq : marker reverse sequences